Etiologia
Qualquer disfunção que resulte em hipocalcemia elevará os níveis do paratormônio (PTH) e poderá causar hiperparatireoidismo secundário (HPTS).[9]
As três principais etiologias que podem levar a esse quadro são: doença renal crônica (DRC), síndromes de má absorção ou exposição solar inadequada crônica.
Na DRC, há perda de 1-alfa-hidroxilase no rim, resultando em redução da conversão de 25-hidroxivitamina D em 1,25-di-hidroxivitamina D ativa. O nível baixo de 1,25-di-hidroxivitamina D, com ou sem hipocalcemia, é detectado pelos receptores da glândula paratireoide e resulta no aumento da secreção de PTH, observado comumente na DRC em estágio avançado (estágios 3 a 5D).
Em afecções como doença de Crohn, doença celíaca, pancreatite crônica, doença de Whipple ou após cirurgia de bypass gástrico ocorre má absorção de gordura, que contribui para a redução da absorção de vitamina D e cálcio alimentar (piorada se houver ingestão inadequada destes), levando, no final, à hipocalcemia e ao aumento do PTH.[2][9]
Para a maioria das pessoas, a exposição à luz solar é a principal fonte de vitamina D.[2] No entanto, as preocupações com o aumento do câncer de pele em decorrência da radiação ultravioleta resultaram em campanhas bem-sucedidas para minimizar a exposição ao sol durante exatamente os horários em que a vitamina D pode ser sintetizada na pele.[10] A exposição inadequada à vitamina D é comum entre pacientes idosos, particularmente naqueles que sempre usam protetor solar, estão incapacitados de sair de casa, institucionalizados ou hospitalizados.[11] Esse fato pode causar deficiência de vitamina D que, por sua vez, pode causar hipocalcemia e consequente aumento da secreção de PTH.
Outras causas incluem:
Baixa ingestão alimentar de cálcio
Perda de cálcio ou necessidade metabólica aumentadas:
Crescimento ósseo no período de crescimento
Gestação e amamentação
Tratamento com bifosfonato
Hipercalciúria idiopática
Diuréticos de alça
Rabdomiólise
Sepse
Redução do efeito do PTH:
DRC
Pseudo-hipoparatireoidismo (deficiência de proteína G)
Fisiopatologia
O metabolismo da vitamina D é essencial para compreender a fisiopatologia das principais causas de HPTS. Há duas formas principais de vitamina D: colecalciferol (vitamina D3) e ergocalciferol (vitamina D2).
[Figure caption and citation for the preceding image starts]: Metabolismo da vitamina DCriado por Dr. Syazrah Salam; usado com permissão [Citation ends].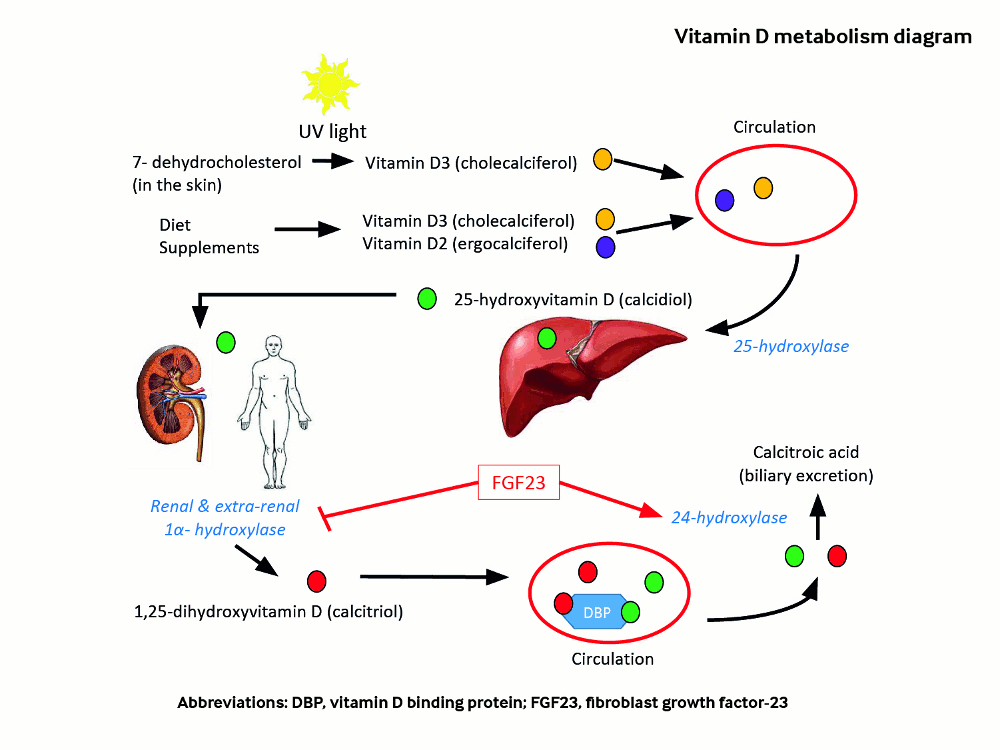
O colecalciferol (vitamina D3) é produzido na pele após exposição solar e é encontrado em alguns poucos alimentos.[12][13] A exposição à luz solar é a principal fonte de vitamina D para a maioria das crianças e adultos.[2] O colecalciferol é naturalmente encontrado em poucos alimentos: salmão, atum, cavala e óleos de fígado de peixe são as melhores fontes, com quantidades menores encontradas em fígado de boi, queijo e gema de ovo. O ergocalciferol (vitamina D2) tem uma cadeia lateral diferente da cadeia lateral do colecalciferol. Ele também é encontrado naturalmente, em pequenas quantidades, em alguns cogumelos.[13] As vitaminas D2 e D3 são usadas para enriquecer leite, pão e em complexos polivitamínicos nos EUA. Na Europa, a vitamina D3 é usada quase exclusivamente em complexos polivitamínicos e enriquecimento de alimentos.
Uma vez que a vitamina D (D2 ou D3) é produzida na pele ou ingerida na alimentação, ela sofre duas hidroxilações, sendo a primeira no fígado para formar a 25-hidroxivitamina D. Esse composto entra na circulação carreado pela proteína de ligação da vitamina D (DBP) e é transportado para o rim, local em que o receptor megalina transfere o complexo DBP-25-hidroxivitamina D até o túbulo renal. Aqui, a enzima 25-hidroxivitamina D 1-alfa-hidroxilase (CYP27B) introduz uma função hidroxila para formar a 1,25-di-hidroxivitamina D.[2]
O fator de crescimento de fibroblastos-23 (FGF-23) é um importante regulador do metabolismo da vitamina D. O FGF-23 inibe a atividade da 1-alfa-hidroxilase nos rins e aumenta a atividade da 24-hidroxilase, que remove o colecalciferol e a 1,25-di-hidroxivitamina D da circulação. O efeito geral é a redução no nível de 1,25-di-hidroxivitamina D.[14]
No intestino, a 1,25-di-hidroxivitamina D induz a expressão de uma proteína de ligação ao cálcio do canal epitelial de cálcio (calbindina) e de diversas outras proteínas que auxiliam no aumento do transporte de cálcio da alimentação para a circulação.[2] A 1,25-di-hidroxivitamina D também interage com o receptor nuclear de vitamina D no osteoblasto, estimulando a expressão do receptor ativador de fator nuclear kappa-B (RANKL), que leva à maturação do osteoclasto e, assim, à reabsorção óssea. Dessa forma, a 1,25-di-hidroxivitamina D auxilia na manutenção da homeostase de cálcio, aumentando a eficiência da absorção intestinal de cálcio e mobilizando depósitos de cálcio do esqueleto.
Qualquer deficiência de vitamina D causa uma diminuição na eficiência da absorção intestinal do cálcio (e fósforo) alimentar.[15] Isso resulta em uma queda transitória do cálcio ionizado, desencadeando imediatamente um aumento na produção e secreção de PTH.[9] O PTH age de modo a aumentar o nível de cálcio ionizado no sangue, interagindo com seu receptor de membrana em osteoblastos maduros, o que, por sua vez, induz a expressão de RANKL. A proteína RANKL é reconhecida por uma proteína chamada ativador de fator nuclear kappa-B (RANK), presente na membrana plasmática de pré-osteoclastos. A interação RANKL-RANK resulta em aumento da produção e maturação de osteoclastos.[2] O PTH diminui também a expressão gênica da osteoprotegerina (um receptor chamariz da RANKL) nos osteoblastos, potencializando a osteoclastogênese. Os osteoclastos liberam ácido clorídrico e colagenases para destruir o osso, resultando na mobilização dos depósitos de cálcio do esqueleto. O HPTS induzido pela deficiência de vitamina D resulta em perda óssea, que pode desencadear e exacerbar a osteoporose.[2]
Outro efeito da deficiência de vitamina D e do HPTS é a perda de fósforo na urina e a diminuição dos níveis de fósforo sérico.[15] O produto cálcio-fósforo inadequado resultante faz com que a matriz óssea formada pelos osteoblastos seja anormalmente mineralizada. Em crianças, o peso corporal faz com que o esqueleto anormalmente mineralizado desenvolva as deformidades raquíticas clássicas, como pernas arqueadas ou joelhos varos. No entanto, em adultos, geralmente há mineralização suficiente para impedir as deformidades do esqueleto, embora, em um estado de deficiência de vitamina D, os osteoides recém-formados não possam ser mineralizados adequadamente, levando à osteomalácia.[15] Isso está associado à dor latejante nos ossos, muitas vezes erroneamente diagnosticada como fibromialgia, miosite ou síndrome da fadiga crônica. Uma possível explicação para a dor nesses pacientes é a de que os osteoides fracamente mineralizados se tornam hidratados e não são capazes de fornecer suporte suficiente para as fibras sensoriais no periósteo.[16] A dor geralmente afeta a pelve, quadris, pernas, coluna lombar e costelas.
Distúrbio ósseo e mineral relacionado à doença renal crônica
O distúrbio ósseo e mineral da doença renal crônica (DRC-DOM) é definido como um distúrbio sistêmico do metabolismo ósseo e mineral decorrente da DRC manifestada por um ou uma combinação dos seguintes fatores:[1]
anormalidades do cálcio, fósforo, paratormônio (PTH) ou metabolismo de vitamina D
anormalidades na renovação óssea, mineralização, volume, crescimento linear ou força
Calcificação vascular ou de outros tecidos moles.
A fisiopatologia do distúrbio ósseo e mineral da doença renal crônica (DRC-DOM) é complexa. Há principalmente um declínio gradual na excreção de fosfato renal total, embora, paradoxalmente, a excreção por néfron individual aumente de forma acentuada, o que mantém o fosfato sérico na faixa normal até a DCR de estágio 4. Há também aumento nos níveis de PTH e FGF-23, dois hormônios fosfatúricos que parecem acelerar o desenvolvimento da doença vascular, especialmente no contexto de DRC.[17] Evidências disso foram fornecidas pelo estudo Framingham Offspring, que demonstrou que a hiperfosfatemia estava associada a aumento do risco de doença cardiovascular mesmo em indivíduos sem DRC, e pela NHANES 3 (Third National Health and Nutrition Examination Survey - terceira pesquisa nacional de avaliação da saúde e nutrição).[18][19] Embora o FGF-23 esteja implicado na fisiopatologia do DRC-DOM, seu uso na prática clínica como um biomarcador de progressão da doença ainda precisa ser testado de forma prospectiva. Há também questões não resolvidas com respeito a testes, e a capacidade de promover uma redução sustentada e significativa nas concentrações de FGF-23 sérico permanece imprecisa.
[Figure caption and citation for the preceding image starts]: Fisiopatologia do hiperparatireoidismo secundário no DRC-DOMCriado pelo BMJ Knowledge Center a partir do fluxograma original do Dr. Syazrah Salam; usado com permissão [Citation ends].
Há um espectro de anormalidades histomorfométricas que pode ser observado em pacientes com DRC, e, até o momento, dados clínicos inadequados e a natureza heterogênea da doença impossibilitam a criação de um sistema de classificação confiável e preciso baseado apenas em anormalidades bioquímicas séricas para auxiliar no diagnóstico e tratamento da DRC.[20] Na verdade, a homeostase mineral óssea disfuncional e a calcificação de tecidos moles não são exclusivas da DRC e são processos multifatoriais que podem apresentar mais de uma etiologia subjacente. Por exemplo, fragilidade óssea, aumento do risco de fratura e calcificação vascular com doença aterosclerótica estão associados a processos normais de envelhecimento e podem ocorrer em pacientes com função renal normal ou apenas levemente reduzida, só existindo concomitantemente com o DRC-DOM quando a função renal está deteriorada. Tradicionalmente, uma biópsia óssea poderia ser justificada na presença de inconsistências bioquímicas, dor óssea inexplicável ou fraturas que não respaldassem o diagnóstico de DRC-DOM.[1] No entanto, a biópsia óssea raramente é realizada na prática clínica atual devido à sua natureza invasiva.
O uso deste conteúdo está sujeito ao nosso aviso legal