Etiologia
A etiologia da arterite de Takayasu é desconhecida. Acredita-se que fatores genéticos e ambientais influenciem o desenvolvimento da doença.[4] Mecanismos de imunidade mediada por células foram implicados.[1] O rastreamento genético demonstrou polimorfismos nos genes interleucina 12 (IL-12), IL-6 e IL-2 em uma população de pacientes turcos com arterite de Takayasu.[15] Há relatos de antígeno leucocitário Bw5 (HLA-Bw5) e antígeno leucocitário B39.2 (HLA-B39.2) com frequência elevada em algumas populações.[16][17]
Fisiopatologia
A arterite de Takayasu é uma vasculite mediada imunologicamente caracterizada pela inflamação granulomatosa das grandes artérias. Mecanismos de imunidade mediada por células foram implicados.[1][4] Acredita-se que a interleucina (IL)-6 e a IL-17 desempenhem um papel importante na patogênese da arterite de Takayasu.[18] Alguns pacientes foram tratados com um inibidor da IL-6 e apresentaram respostas favoráveis.[19][20]
A resposta imunológica e inflamatória observada nas artérias é semelhante à observada nas artérias grandes da arterite de células gigantes.[1][4] Durante a fase aguda da vasculite, a inflamação começa nos vasa vasora da adventícia das artérias musculares.[1][9] As células T são proeminentes na resposta celular inicial e também pode haver o envolvimento de anticorpos anti-células endoteliais.[1][4][21][22][Figure caption and citation for the preceding image starts]: A fotomicrografia da aorta de um paciente com arterite de Takayasu revela marcante espessamento da camada intimal, infiltrados inflamatórios na média e necrose laminarUsado com permissão do acervo de Dylan Miller, MD, Mayo Clinic [Citation ends].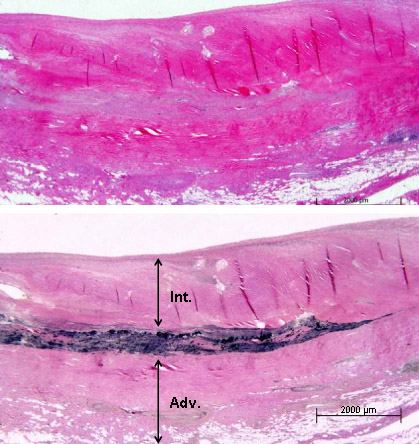
Classificação
2012 International Chapel Hill Consensus Conference on the Nomenclature of Vasculitides[5]
Categoriza a vasculite baseada no tipo predominante de vasos envolvidos e outras características incluindo etiologia, patogênese, tipo de inflamação, distribuição de órgãos preferível, manifestações clínicas, predisposições genéticas e características demográficas distintas.
Vasculite dos grandes vasos
Arterite de Takayasu
Arterite de células gigantes
Vasculite de vasos médios
Poliarterite nodosa
Doença de Kawasaki
Vasculite dos pequenos vasos
Vasculite associada ao ANCA (anticorpo anticitoplasma de neutrófilo)
Poliangiite microscópica
Granulomatose com poliangiite (anteriormente conhecida como granulomatose de Wegener)
Granulomatose eosinofílica com poliangiite (Churg-Strauss)
Vasculite por imunocomplexo
Doença por anticorpo antimembrana basal glomerular (anti-GBM)
Vasculite crioglobulinêmica
Vasculite de imunoglobulina A (Henoch-Schönlein)
Vasculite urticariforme hipocomplementêmica
Vasculite de vasos variáveis
Síndrome de Behçet
Síndrome de Cogan
Vasculite de órgão único
Vasculite associada a doença sistêmica
Vasculite associada a etiologia provável
Classificação angiográfica da arterite de Takayasu[6]
A classificação baseia-se nos vasos envolvidos no processo inflamatório conforme observado por angiografia.
Tipo I: ramos do arco aórtico
Tipo IIa: aorta ascendente, arco aórtico e ramos do arco aórtico
Tipo IIb: aorta ascendente, arco aórtico e seus ramos e aorta descendente torácica
Tipo III: aorta ascendente torácica, aorta abdominal e/ou artérias renais
Tipo IV: aorta abdominal e/ou artérias renais
Tipo V: características dos tipos IIb e IV
O uso deste conteúdo está sujeito ao nosso aviso legal