Pruebas diagnósticas
Primeras pruebas diagnósticas para solicitar
IRM cerebral
Prueba
La resonancia magnética (IRM) debe solicitarse, tan pronto como exista una sospecha de demencia rápidamente progresiva y debe incluir secuencias de T1, T2, imágenes ponderadas por difusión (DWI), mapa del coeficiente de difusión atenuado (ADC) y recuperación de inversión atenuada por líquido (FLAIR).[76] Si es posible, las secuencias DWI y ADC se deben obtener en los planos coronal y axial para minimizar el artefacto en la interfaz cerebro-aire.[Figure caption and citation for the preceding image starts]: Cambios en los núcleos basales observados en la enfermedad de Creutzfeldt-Jakob. (A) La IRM con recuperación de inversión atenuada por líquido y (B) la IRM ponderada en difusión del mismo paciente demuestran hiperintensidades en los núcleos basales bilaterales (flechas). También se observa hiperintensidad leve bilateral en los núcleos pulvinar y medial del tálamoDe la colección personal del Dr. M. Geschwind [Citation ends].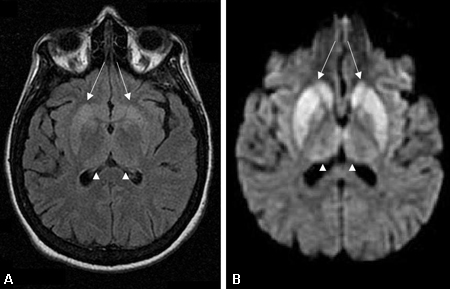 [Figure caption and citation for the preceding image starts]: Hiperintensidad bilateral en los núcleos pulvinar y medial del tálamo (puntas de flecha) en (A) una IRM con recuperación de inversión atenuada por líquido y (B) una IRM ponderada en difusión en un paciente con la enfermedad de Creutzfeldt-Jakob. Este paciente también presenta hiperintensidad significativa en los núcleos basales en las dos secuencias (flechas)De la colección personal del Dr. M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Hiperintensidad bilateral en los núcleos pulvinar y medial del tálamo (puntas de flecha) en (A) una IRM con recuperación de inversión atenuada por líquido y (B) una IRM ponderada en difusión en un paciente con la enfermedad de Creutzfeldt-Jakob. Este paciente también presenta hiperintensidad significativa en los núcleos basales en las dos secuencias (flechas)De la colección personal del Dr. M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Ribete cortical difuso (flechas) observado en una IRM con imágenes ponderadas en difusión (DWI) y en menor grado en una IRM con recuperación de inversión atenuada por líquido (FLAIR). Las dos secuencias muestran hiperintensidades en las circunvoluciones de la corteza cerebralDe la colección personal del Dr. M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Ribete cortical difuso (flechas) observado en una IRM con imágenes ponderadas en difusión (DWI) y en menor grado en una IRM con recuperación de inversión atenuada por líquido (FLAIR). Las dos secuencias muestran hiperintensidades en las circunvoluciones de la corteza cerebralDe la colección personal del Dr. M. Geschwind [Citation ends].
En general, las alteraciones halladas en las secuencias ADC y DWI de la IRM, en la enfermedad de Creutzfeldt-Jakob esporádica (ECJe) y la variante ECJ (vECJ), no se han informado en otras demencias similares y sugieren en gran medida la presencia de ECJ.[67]
Si las exploraciones son de buena calidad, la IRM, las DWI y la FLAIR pueden presentar una sensibilidad de un 92 a 96% y una especificidad de un 93 a 94%.[65][67][77]
En las imágenes T1 con gadolinio, la ECJ generalmente no muestra un realce por contraste, ni alteraciones en la intensidad de señal de la sustancia blanca; si estos llegan a detectarse, se deben investigar otros diagnósticos.[78][79]
Se ha informado hipointensidad en T1 en el globus pallidus (globo pálido) en la ECJ.[87] En la vECJ, se puede observar el signo pulvinar, un término que hace referencia a la hiperintensidad pulvinar bilateral. Cuando los núcleos pulvinar y medial del tálamo muestran intensidad en la señal, se debe sospechar la vECJ.[71][80][81] Ciertas formas de ECJ genética y otros tipos de enfermedades priónicas genéticas (mutaciones particulares) presentan hallazgos en la IRM similares a los observados en la IRM de difusión (DWI) en la ECJe, aunque muchas enfermedades priónicas genéticas solo muestran atrofia.
Resultado
a menudo demuestra hiperintensidad en la corteza cerebral (ribete cortical), los núcleos basales (núcleo caudado y putamen) y el tálamo en las imágenes ponderadas en difusión (DWI), en las secuencias de recuperación de inversión atenuada por líquido (FLAIR) e hipointensidad (difusión restricta) en las secuencias del mapa de coeficiente de difusión atenuado (ADC)
electroencefalograma (EEG)
Prueba
Las alteraciones, a pesar de ser moderadamente específicas, solo tienen una sensibilidad de aproximadamente un 60% y es posible que no aparezcan hasta las últimas etapas de la enfermedad.[14][82]
Una vez que se hayan descartado otras afecciones con alteraciones similares en el EEG, estos hallazgos pueden resultar altamente específicas para las enfermedades priónicas.[71]
Resultado
desaceleración generalizada, focal o difusa, y complejos periódicos de ondas puntiagudas
Pruebas diagnósticas que deben considerarse
conversión inducida por temblores (QuIC)
Prueba
Prueba sensible y específica que detecta la isoforma de la proteína priónica asociada a la enfermedad en el líquido cefalorraquídeo (LCR) de pacientes con enfermedad de Creutzfeldt-Jakob (ECJ) esporádica.[68] La conversión inducida por el temblor (QuIC) en tiempo real (RT-QuIC) y la QuIC de criterio de valoración (EP-QuIC) son las versiones que actualmente utilizadas por los laboratorios de diagnóstico, según el país.
Resultado
positiva
biomarcadores en el LCR
Prueba
A pesar de que los biomarcadores en el LCR son útiles para la confirmación del rápido deterioro neuronal, no pueden diagnosticar ni descartar, de forma definitiva, las enfermedades priónicas.[83][88][89][90] Estas pruebas deben interpretarse con precaución dado que su sensibilidad y su especificidad para la enfermedad de Creutzfeldt-Jakob esporádica (ECJe) aún no están claras.
El nivel de la proteína Abeta-42 puede disminuir y los niveles de las proteínas tau total (T-tau) y tau fosforilada (P-tau) pueden ser elevados en la enfermedad de Alzheimer.
El nivel de la proteína 14-3-3 hallado en el LCR se ha informado como un fuerte indicador de ECJ. Sin embargo, la sensibilidad y la especificidad de esta prueba varían considerablemente en la literatura.[71][91][92][93] Las guías de práctica clínica actuales se están alejando del uso de esta prueba.
Algunos estudios sugieren que las proteínas en el LCR, como las proteínas 14-3-3, tau total y la enolasa neuroespecífica, presentan una mayor sensibilidad en las enfermedades rápidamente progresivas, lo que sustenta la idea de que se liberan durante la lesión y la muerte neuronal y no son necesariamente específicas de las enfermedades priónicas.[71][84][88][89]
Resultado
la proteína 14-3-3 (p. ej., western blot) puede ser positiva; la proteína tau total (T-tau) elevada ( >1200 picogramos/mL); la enolasa neuroespecífica elevada (>35 nanogramos/mL)
pruebas genéticas en el gen de la proteína priónica
Prueba
El inicio y las manifestaciones clínicas de todas las formas de enfermedades priónicas se ven muy afectadas por el codón polimórfico 129 del gen de la proteína priónica endógena (PRNP). El codón 129 puede ser metionina (M) o valina (V). Las combinaciones homocigóticas (p. ej., MM o VV) traen como consecuencia un riesgo más alto de desarrollar enfermedades priónicas. El tipo de prión (tipo 1 o 2) también influye en la presentación de la enfermedad; sin embargo, la tipificación de priones solo se puede determinar a partir de tejido cerebral congelado obtenido mediante biopsia cerebral o autopsia.[14][23]
Las enfermedades priónicas genéticas se producen por una mutación en el gen que codifica la proteína priónica PRNP, ubicado en el cromosoma 20.[6] Hasta la fecha se han identificado más de 40 mutaciones diferentes en el gen PRNP, cada una presentándose con su propio fenotipo de enfermedad (p. ej., ECJ hereditaria, Gerstmann-Straussler-Scheinker e insomnio familiar fatal).
Las mutaciones en el gen PRNP se transmiten de manera autosómica dominante.[6] Es importante destacar que alrededor del 60% de los pacientes con enfermedad priónica de origen genético no tenían antecedentes familiares positivos de enfermedad priónica. Un examen más detenido suele revelar antecedentes familiares de Alzheimer o Parkinson que probablemente fueron mal diagnosticados, o la muerte de uno de los progenitores antes de la aparición de los síntomas.[21][22]
Los pacientes y sus familias también deben recibir asesoramiento genético y que comprendan las consecuencias implícitas antes de conocer tales resultados. Dado que las enfermedades priónicas genéticas son autosómicas dominantes, generalmente se utiliza el protocolo utilizado para la enfermedad de Huntington.[86] Este protocolo se utiliza para muchos trastornos neurológicos autosómicos dominantes a fin de asegurarse que los pacientes, sus familias y otros comprendan las implicaciones psicológicas, psiquiátricas, médicas, legales y otras implicaciones de las pruebas genéticas.
Las muestras deben estar acompañadas por breves antecedentes clínicos y se deben enviar el mismo día de su recolección.
The National Creutzfeldt-Jakob Disease Research and Surveillance Unit Opens in new window Public Health Agency of Canada: prion disease information Opens in new window National Prion Disease Pathology Surveillance Center Opens in new window
Resultado
positiva
biopsia (cerebro, amígdala)
Prueba
La biopsia de cerebro previa a la muerte es la única forma de diagnóstico definitivo de la enfermedad priónica esporádica. En la variante ECJ, se puede utilizar una biopsia de amígdala para el diagnóstico.
Dado que la acumulación de proteína en el cerebro presenta un patrón impredecible, se puede obtener un resultado falso negativo.
Los procedimientos de la biopsia de cerebro pueden implicar un riesgo innecesario de infección o un daño cerebral posterior para los pacientes. Aunque se haya confirmado el diagnóstico aún no existe un tratamiento disponible.
Las proteínas priónicas son resistentes a los métodos de esterilización quirúrgica estándar, y el personal médico que realiza el procedimiento puede estar en riesgo de contagio de la ECJ. Se deben seguir los protocolos adecuados para minimizar el riesgo de transmisión.[27]
En definitiva, no se recomienda la realización de la biopsia de cerebro para el diagnóstico de la ECJ, especialmente dado que, hoy en día, los antecedentes clínicos, los estudios por imagen cuantitativa QuIC y la IRM son útiles para tal fin.[67][71]
Si se realiza una biopsia de cerebro, debe obtenerse tejido de las áreas en las que la IRM indica la presencia de alteraciones, donde sea posible.
A pesar de que los cambios microscópicos característicos son típicos de esta enfermedad, otras afecciones también pueden presentar estos mismos cambios microscópicos.
La presencia del prión patogénico (PrPSc) por inmunohistoquímica o western blot es necesaria para el diagnóstico definitivo.[3][36]
En aproximadamente el 10% de los casos de ECJ, se detectan placas amiloides de PrPSc y, en casos de Gerstmann-Straussler-Scheinker, un núcleo amiloide de PrPSc se encuentra rodeado por otro grupo de glóbulos amiloides más pequeños.
La variante ECJ (vECJ) también presenta una característica patológica relativamente única de placas amiloides de PrPSc en el núcleo rodeadas por vacuolas. Estas se denominan placas floridas dado que se cree que se parecen a una flor.
En la vECJ las PrPSc también pueden identificarse en el sistema linforreticular durante la evolución de la enfermedad y las biopsias de amígdala pueden mostrar la presencia de PrPSc, aunque no en otras formas de enfermedades priónicas en seres humanos.[3][37]
Resultado
microscópico: vacuolación (cambios espongiformes), pérdida neuronal y astrogliosis; histológico: presencia de prión patógeno (PrPSc) por inmunohistoquímica o western blot; puede mostrar amiloide
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad