Abordaje
La presentación de los pacientes puede ser muy útil para determinar si los síntomas coinciden con los de la enfermedad de Creutzfeldt-Jakob (ECJ) o si se pueden atribuir a otra afección. Se debe realizar un diagnóstico de exclusión en los pacientes, aunque los hallazgos de la IRM cerebral con imágenes ponderadas en difusión (DWI) y mapa de coeficiente de difusión atenuado (ADC) resultan muy útiles para el diagnóstico.[65][66][67] La conversión en tiempo real inducida por el temblor (RT-QuIC) del LCR también posee un alto valor diagnóstico.[68][69][70]
Antecedentes
El inicio de la ECJ esporádica (ECJe) es subagudo, aunque el avance general es bastante rápido con relación a otras demencias de desarrollo más lento como la enfermedad de Alzheimer. Se deben observar posibles factores de riesgo que incluyen ciertos procedimientos médicos a los que el paciente puede haberse sometido (ECJ iatrogénica) y detalles de viajes al extranjero (fechas, duración, consumo de carne vacuna), especialmente en el Reino Unido para la variante ECJ (ECJv).
Se desconocía la existencia de antecedentes familiares de enfermedades priónicas en más del 60% de los pacientes con ECJ en quienes se halló una mutación en el gen de la proteína priónica (PRNP). La recopilación de un árbol genealógico detallado es importante, pero se recomienda que todos los pacientes se sometan a pruebas de mutación del gen, incluso si no hay antecedentes familiares. Se deben evaluar los antecedentes familiares de demencia, otras afecciones neurológicas y trastornos psiquiátricos, dado que las formas genéticas de las enfermedades priónicas a menudo se diagnostican incorrectamente como otras enfermedades.
Exploración física y neurológica
La presentación clínica de las enfermedades priónicas probablemente dependa de las regiones del cerebro en las que se acumulan los priones. Los síntomas pueden imitar a otras enfermedades neurológicas o psiquiátricas. Los pacientes con sospecha de enfermedad priónica se deben derivar de inmediato a un neurólogo. Es esencial realizar un examen neurológico detallado. Los signos hallados en el examen ayudarán a dirigir el análisis diagnóstico de exclusión.
Lo más frecuente es que la ECJe se produzca durante la séptima década de la vida (pacientes con los 60 años), y tipicamente presenta con lo siguiente.
Alteraciones cognitivas, falta de coordinación, cambios conductuales y/o visuales.[71]
En un estudio de más de 100 casos de ECJ, los problemas cognitivos fueron el primer síntoma más frecuente.[71] Los pacientes padecen con frecuencia pérdida de la memoria, afasia y dificultades con el funcionamiento ejecutivo (p. ej., capacidad de organizar, planificar y realizar múltiples tareas).
Los siguientes síntomas más frecuentes fueron del cerebelo, conductuales y otros síntomas motores. Los signos motores incluyen parkinsonismo, mioclonía y ataxia de la marcha o los miembros. La afectación de los lóbulos frontales o las conexiones subcorticales del lóbulo frontal pueden afectar la conducta, causando agitación, depresión u otros signos psiquiátricos.[72]
Algunos pacientes también pueden describir síntomas inespecíficos o constitucionales, como vértigo, cefalea y mareos que pueden preceder a la enfermedad en semanas o incluso meses.[73]
Los síntomas visuales son generalmente menos frecuentes, pero pueden incluir diplopia, alucinaciones y otras distorsiones visuales.[72]
La vECJ se manifiesta de manera bastante diferente que la de la forma esporádica.
Por lo general, afecta a adultos jóvenes y adolescentes.
En la mayoría de los pacientes, los primeros síntomas son psiquiátricos e incluyen depresión profunda y deterioro cognitivo leve.
Posteriormente en la evolución, los pacientes desarrollan demencia, ataxia, síntomas sensoriales dolorosos y/o un trastorno del movimiento.[11][21][71]
La ECJ genética es causada por más de 40 mutaciones diferentes en el gen de la proteína priónica (PRNP) y luego puede subdividirse en ECJ hereditaria, Gerstmann-Straussler-Scheinker (GSS) e insomnio familiar fatal (IFF), en función de los hallazgos clínicos y patológicos.
La ECJ hereditaria puede tener una evolución más prolongada y más lenta que la GSS o que el IFF.
La evolución clínica de la GSS es generalmente más prolongada y lenta que la de la ECJe, la que a menudo dura unos años y tanto como una década. El parkinsonismo o la ataxia pueden ser los signos que presentan la GSS y que pueden diagnosticarse incorrectamente como otras enfermedades neurodegenerativas más lentas, como las demencias parkinsonianas atípicas, la enfermedad de Parkinson y la atrofia multisistémica.[74]
El IFF generalmente se presenta como un síndrome de insomnio y disautonomía. Puede presentarse ataxia o descoordinación cerebelosa. La demencia se manifiesta en una etapa posterior de la evolución de la enfermedad.[75]
resonancia magnética (IRM)
Se debe indicar una IRM apenas se sospeche de una demencia rápidamente progresiva. Los hallazgos son especialmente valiosos en el diagnóstico de las enfermedades priónicas y tienen una sensibilidad y una especificidad elevadas cuando se utiliza la recuperación de inversión atenuada por líquido (FLAIR) y, particularmente, imágenes ponderadas en difusión (DWI) o mapa de coeficiente de difusión atenuado (ADC).[66][67][76][Figure caption and citation for the preceding image starts]: Cambios en los núcleos basales observados en la enfermedad de Creutzfeldt-Jakob. (A) La IRM con recuperación de inversión atenuada por líquido y (B) la IRM ponderada en difusión del mismo paciente demuestran hiperintensidades en los núcleos basales bilaterales (flechas). También se observa hiperintensidad leve bilateral en los núcleos pulvinar y medial del tálamoDe la colección personal del Dr. M. Geschwind [Citation ends].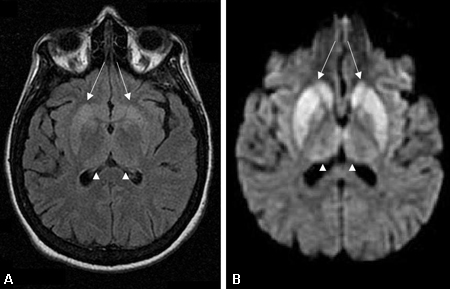 [Figure caption and citation for the preceding image starts]: Hiperintensidad bilateral en los núcleos pulvinar y medial del tálamo (puntas de flecha) en (A) una IRM con recuperación de inversión atenuada por líquido y (B) una IRM ponderada en difusión en un paciente con la enfermedad de Creutzfeldt-Jakob. Este paciente también presenta hiperintensidad significativa en los núcleos basales en las dos secuencias (flechas)De la colección personal del Dr. M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Hiperintensidad bilateral en los núcleos pulvinar y medial del tálamo (puntas de flecha) en (A) una IRM con recuperación de inversión atenuada por líquido y (B) una IRM ponderada en difusión en un paciente con la enfermedad de Creutzfeldt-Jakob. Este paciente también presenta hiperintensidad significativa en los núcleos basales en las dos secuencias (flechas)De la colección personal del Dr. M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Ribete cortical difuso (flechas) observado en una IRM con imágenes ponderadas en difusión (DWI) y en menor grado en una IRM con recuperación de inversión atenuada por líquido (FLAIR). Las dos secuencias muestran hiperintensidades en las circunvoluciones de la corteza cerebralDe la colección personal del Dr. M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Ribete cortical difuso (flechas) observado en una IRM con imágenes ponderadas en difusión (DWI) y en menor grado en una IRM con recuperación de inversión atenuada por líquido (FLAIR). Las dos secuencias muestran hiperintensidades en las circunvoluciones de la corteza cerebralDe la colección personal del Dr. M. Geschwind [Citation ends].
La IRM de DWI y FLAIR muestra hiperintensidad en la sustancia gris de las circunvoluciones de la corteza cerebral, los núcleos basales (caudado y putamen) y, con menos frecuencia, en el tálamo.[67][77]
En las imágenes T1 con gadolinio, la ECJ generalmente no muestra un realce por contraste, ni alteraciones en la intensidad de señal de la sustancia blanca. Si estos llegan a detectarse, se deben investigar otros diagnósticos.[78][79] Algunos pacientes con ECJ presentan hiperintensidad en T1 en el globus pallidus (globo pálido).
En la vECJ se puede observar el signo del pulvinar, un término que hace referencia a la hiperintensidad bilateral del núcleo pulvinar. Cuando los núcleos pulvinar y medial del tálamo muestran intensidad en la señal, se debe sospechar la vECJ.[71][80][81]
En general, las alteraciones halladas en las secuencias ADC y DWI de la IRM en la ECJe y la vECJ no se han informado en otras demencias similares y sugieren en gran medida la presencia de ECJ.[67]
electroencefalograma (EEG)
El EEG es un estudio de rutina. Los hallazgos pueden incluir desaceleración generalizada, focal o difusa, y complejos periódicos de ondas puntiagudas. Estas alteraciones, a pesar de ser moderadamente específicas, solo tienen una sensibilidad de aproximadamente un 60% y es posible que no aparezcan hasta las últimas etapas de la enfermedad.[77][82] Una vez que se hayan descartado otras afecciones con alteraciones similares en el EEG, estos hallazgos pueden resultar altamente específicas para las enfermedades priónicas.[71]
Prueba de LCR
La conversión inducida por el temblor en tiempo real (RT-QuIC) se está utilizando en varios países como ensayo directo para detectar la presencia de priones anormales en el líquido cefalorraquídeo (LCR). Presenta una especificidad y sensibilidad muy alta a la enfermedad de Creutzfeldt-Jakob (ECJ), en particular las formas esporádicas.[68][69][70]
Se ha informado el nivel de la proteína 14-3-3 encontrado en el líquido cefalorraquídeo (LCR) como un fuerte indicador de la enfermedad de Creutzfeldt-Jakob (ECJ); sin embargo, la sensibilidad y la especificidad de esta prueba varían considerablemente según la fuente.[71] Mientras que existe un gran desacuerdo respecto de la sensibilidad de esta prueba para la ECJe, cada vez cobra mayor aceptación en la comunidad de la neurología que esta prueba no es lo suficientemente específica para la ECJe u otras enfermedades priónicas en seres humanos.[83] Las guías de práctica clínica actuales se están alejando del uso de esta prueba.
En estudios de investigación, se ha sugerido que las proteínas en el LCR, como la proteína tau total (T-tau) y la enolasa neuroespecífica (ENE), presentan una sensibilidad equivalente o ligeramente más elevada aunque una especificidad mucho más alta que la 14-3-3. Sin embargo, estas proteínas también pueden aumentar en otras enfermedades no priónicas rápidamente progresivas, lo que sustenta la idea de que se liberan durante la lesión y muerte neuronal y que no son necesariamente específicas de las enfermedades priónicas.[84]
A pesar de que son útiles para la confirmación del rápido deterioro neuronal, estos biomarcadores no pueden diagnosticar ni descartar en forma definitiva las enfermedades priónicas.[66][71][83][85]
Análisis de sangre y pruebas genéticas
No se dispone de análisis de sangre para la detección de priones, aunque se debe realizar el cribado de pacientes para las mutaciones genéticas en el gen PRNP.
Las muestras se deben enviar a un laboratorio especializado.
The National Creutzfeldt-Jakob Disease Research and Surveillance Unit Opens in new window
Public Health Agency of Canada: prion disease information Opens in new window
National Prion Disease Pathology Surveillance Center Opens in new window
Se recomienda que los pacientes y sus familias reciban asesoramiento genético y que comprendan las consecuencias antes de someterse a las pruebas y de conocer los resultados genéticos. Generalmente se utiliza el protocolo de asesoramiento genético utilizado para las pruebas para la enfermedad de Huntington en las pruebas para el PRNP.[86]
Biopsia
La biopsia de cerebro previa a la muerte es la única forma de diagnóstico definitivo de la enfermedad priónica esporádica. En la variante ECJ, se puede utilizar una biopsia de amígdala para el diagnóstico.
Dado que la acumulación de proteína en el cerebro presenta un patrón impredecible, se puede obtener un resultado falso negativo en la biopsia de cerebro. El procedimiento puede implicar un riesgo innecesario de infección o un daño cerebral posterior para los pacientes. Aunque se haya confirmado el diagnóstico todavía no existe un tratamiento disponible.
Las proteínas priónicas también son resistentes a métodos de esterilización quirúrgica estándares, y el personal médico que realiza el procedimiento puede estar en riesgo de contagio de la ECJ. Hay orientación disponible del Department of Health del Reino Unido para minimizar el riesgo de transmisión.[27] La biopsia de cerebro solo se recomienda cuando la IRM es negativa para la ECJ y se han descartado todas las demás enfermedades mediante otros métodos menos invasivos.[71]
Autopsia
La autopsia es altamente recomendada, dado que la confirmación patológica es la única manera de diagnosticar de forma definitiva las enfermedades priónicas, además de la biopsia.[36]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad