Abordagem
A abordagem para um neonato com genitália atípica envolve uma equipe multidisciplinar de subespecialistas pediátricos.[18] Geralmente, inclui um endocrinologista, um neonatologista, um cirurgião urológico, um psicólogo clínico e um profissional de enfermagem especializado. A equipe multidisciplinar principal deve estar vinculada a uma equipe mais ampla, que inclua especialistas em genética, bioquímica, especialidades médicas adultas relevantes, como ginecologia e endocrinologia, serviços sociais e, se possível, um fórum de ética clínica.[8] A comunicação contínua com a família é crucial, bem como a comunicação com o médico de atenção primária.[8][19] Os conceitos a seguir devem orientar a avaliação diagnóstica de neonatos com genitália atípica:[1][8][20]
Atribuição do sexo. O sexo não é atribuído até que a avaliação tenha sido concluída. A criança é chamada de "bebê", não de menino ou menina. A família deve ser incentivada a esperar para dar um nome ao bebê até que o sexo tenha sido atribuído. A atribuição do sexo pode ser realizada mesmo que um diagnóstico endócrino ou genético subjacente não esteja aparente.[21]
Após a avaliação e as investigações iniciais, o bebê deve ser avaliado em um centro com experiência multidisciplinar na avaliação de neonatos com genitália atípica. Em algumas situações, o atendimento em um hospital local pode ser apropriado, com consulta inicial a uma equipe multidisciplinar especializada, em um centro regional.
O ideal é que as discussões com a família sejam conduzidas por um único profissional da equipe, geralmente o endocrinologista. É útil obter a concordância dos pais para discutir os resultados iniciais das investigações de maneira coletiva, em vez de informá-los do resultado de cada exame, pois eles podem colocar uma ênfase excessiva no resultado do nível de testosterona ou cariótipo e não compreender a medida em que deve ser interpretado diante dos resultados de outras investigações.[21]
As necessidades, o histórico, a cultura e as expectativas dos pais devem ser compreendidos e respeitados. Os pais devem tranquilizados e entender que filhos com um distúrbio do desenvolvimento sexual (DDS) podem ter uma vida bem-sucedida e conviver bem na sociedade. As decisões sobre a atribuição do sexo são tomadas pela equipe multidisciplinar de DDS, em conjunto com os pais. Será traçado um plano de manejo para a alta. Também é importante orientar os pais quanto ao desenvolvimento sexual esperado, pois isso os ajuda a prever os problemas com antecedência.[21]
História
O histórico familiar pode identificar membros da família com problemas semelhantes que indiquem uma herança autossômica recessiva ou ligada ao cromossomo X. Isso inclui uma investigação sobre o histórico de consanguinidade, cirurgia genital, infertilidade, virilização materna durante a gestação (por exemplo, alteração da voz ou hirsutismo), virilização de menina na puberdade (deficiência de 5-alfa redutase) ou outras crianças nascidas com genitália atípica. A morte neonatal não explicada de um membro da família pode indicar DDS não diagnosticado associado a insuficiência adrenal.
A maioria das causas genéticas de genitália atípica ocorre por uma condição autossômica recessiva, como hiperplasia adrenal congênita (HAC), deficiência de 5-alfa redutase e defeitos na biossíntese de testosterona. A herança recessiva ligada ao cromossomo X pode sugerir insensibilidade a androgênios (o gene do receptor de androgênio está no cromossomo X). A anamnese pré-natal deve incluir perguntas relacionadas a exposições maternas a androgênios ou medicamentos e sinais de virilização na mãe durante a gestação.
Exame físico
Embora distúrbios diferentes possam se manifestar com achados semelhantes no exame físico, muitas vezes existem aspectos do exame físico que são cruciais e ajudarão a orientar as investigações iniciais.
Exame físico geral: como algumas causas de DDS estão associadas à insuficiência adrenal, é importante confirmar se os sinais vitais (pressão arterial, enchimento capilar e frequência cardíaca) e a glicose sanguínea estão normais, para possibilitar o monitoramento adequado. A presença de características dismórficas e/ou outras anomalias congênitas pode sugerir uma síndrome que inclui genitália atípica (por exemplo, síndrome de Smith-Lemli-Opitz).
Gônadas: a presença de uma ou duas gônadas descarta o DDS 46,XX secundário à deficiência de 21-hidroxilase. Em casos raros, o DDS ovotesticular 46,XX pode apresentar-se com gônadas inguinais. É mais provável que a presença de duas gônadas seja causada por um DDS 46,XY. A presença de apenas uma gônada palpável também pode sugerir o diagnóstico de disgenesia gonadal mista.
Comprimento do pênis: para meninos nascidos a termo, o comprimento do pênis esticado varia de 2.5 cm a 4.5 cm, e para meninas nascidas a termo, o comprimento do clitóris varia de 0.2 cm a 0.85 cm, com variações entre grupos étnicos.[21]
Os neonatos com DDS 46,XX devido à deficiência de 21-hidroxilase podem apresentar dobras labioescrotais hiperpigmentadas
O escore de masculinização externa pode ser um adjuvante útil para o exame clínico, agindo como um guia para o médico local.[22][23] O escore da genitália externa é uma alternativa neutra em termos de gênero, que tem alto grau de correlação com o escore de masculinização externa original.[8][24][Figure caption and citation for the preceding image starts]: Taxa de masculinização externaCriada pelo BMJ Knowledge Centre com base em Davies JH, Cheetham T. Recognition and assessment of atypical and ambiguous genitalia in the newborn. Arch Dis Child. Abril de 2017 [publicação eletrônica antes da impressa]. [Citation ends].
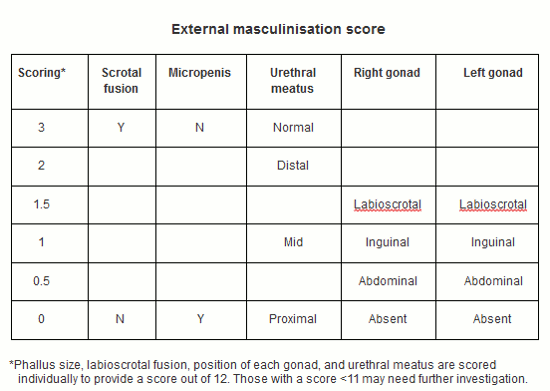
Abertura uretral: a hipospadia ocorre quando não há abertura uretral na ponta do pênis. A hipospadia associada à separação de sacos escrotais ou testículos não descidos pode sugerir um DDS subjacente. Se a abertura uretral estiver na base do pênis, ela poderá ser um seio urogenital em uma mulher virilizada. Isso ocorre quando as aberturas uretral e vaginal estão conectadas internamente e têm saída no períneo através de uma abertura em comum.
Outros sinais a serem observados incluem:[21]
Defeitos na linha média (por exemplo, fenda palatina)
Pigmentação excessiva (por exemplo, pigmentação da aréola)
Anormalidades cardíacas ou esqueléticas.
Investigações adicionais sobre a DDS devem ser consideradas em lactentes com micropênis isolado, clitoromegalia isolada, hipospádia perineal isolada ou qualquer hipospádia familiar, criptorquidia bilateral isolada e naqueles com escore de masculinização externa inferior a 11 (ou escore de genitália externa inferior a 10.5). A presença de um distúrbio metabólico, características dismórficas ou história familiar relevante deve diminuir o limiar para uma investigação mais aprofundada.[8]
Principais quadros clínicos
Gônadas bilaterais impalpáveis em um bebê aparentemente do sexo masculino
Um neonato com gônadas bilaterais impalpáveis, com fenótipo masculino externo, pode na verdade ter cariótipo 46,XX com aumento clitoriano causado pela produção excessiva de androgênio no útero. Esses bebês não devem receber alta até que a hiperplasia adrenal congênita (HAC) secundária à deficiência de 21-hidroxilase tenha sido excluída.
Hérnia inguinal em um bebê aparentemente do sexo feminino
O neonato ainda pode ter um cariótipo 46,XX, e essa apresentação pode ocorrer em, por exemplo, insensibilidade completa a andrógenos, deficiência de 17 beta-hidroxiesteroide desidrogenase e deficiência de 5 alfa-redutase.
Genitália assimétrica
O mosaicismo do cromossomo sexual (por exemplo, 45,X/46,XY) pode causar genitália assimétrica. Na laparoscopia, uma gônada estriada e hemiútero costumam ser identificados no lado contralateral à gônada palpável ou descendente.
Micropênis
Para um menino nascido a termo, o comprimento normal do pênis esticado varia de 2.5 a 4.5 cm.[21] Um comprimento de <2.5 cm é considerado anormal em um bebê a termo do sexo masculino, sobretudo na presença de criptorquidia ou outras anormalidades.[21] O tamanho normal do pênis varia entre os grupos étnicos.
Em bebês do sexo masculino com nascimento pré-termo, o pênis é mais curto, e o comprimento deve ser plotado em um gráfico percentual disponível para bebês nessas condições.[25]
As disfunções na biossíntese de testosterona ou na ação da testosterona podem causar o micropênis (por exemplo, insensibilidade parcial ao andrógeno e deficiência de 5 alfa-redutase).
Aumento clitoriano
O comprimento normal do clitóris em bebês do sexo feminino nascidas a termo varia de 0.2 cm a 0.85 cm, com variações entre grupos étnicos.[21] A hiperplasia adrenal congênita (HAC) secundária à deficiência de 21-hidroxilase é a causa mais comum de cliteromegalia.
Hipospádia
A hipospádia grave pode estar associada à genitália atípica. A hipospádia grave pode ter uma causa genética subjacente em 40% dos casos, inclusive insensibilidade parcial ao androgênio e deficiência de 5 alfa-redutase.[22][26] O neonato com hipospadia, mas com gônadas impalpáveis, tem HAC secundária à deficiência de 21-hidroxilase, até prova em contrário.
Abordagem para um paciente com genitália atípica
Após realizar um histórico e exame físico (que deve incluir a palpação das gônadas) detalhados, o próximo passo é organizar as investigações específicas. Na apresentação inicial, deve-se realizar o monitoramento de eletrólitos regulares, concentrações de glicose antes da ingestão de alimentos e pressão arterial. Deve ser solicitada uma análise cromossômica urgente (cariótipo), com exames de imagem nas primeiras 48 horas e estudos hormonais 48 horas após o nascimento. Em bebês com gônadas impalpáveis, um 17-OHP e outros estudos hormonais devem ser realizados 48 horas após o nascimento. Uma ultrassonografia é útil para definir a presença de útero (estrutura mülleriana), a presença de glândulas adrenais e a morfologia das gônadas. Também deve-se solicitar uma razão de proteína-creatinina na urina.
Análise cromossômica
Na avaliação inicial, uma análise cromossômica deverá ser realizada com no mínimo 30 metáfases para avaliar se há mosaicismo. Os resultados geralmente podem ser obtidos dentro de 72 horas. Um cariótipo formal pode levar dias para ser realizado, e é possível obter um resultado mais rápido solicitando-se uma hibridização in situ fluorescente (FISH) ou uma reação em cadeia da polimerase quantitativa do gene da região determinante do sexo Y (SRY). Outros estudos genéticos poderão ser realizados quando um diagnóstico específico estiver em consideração.[9]
Ensaios de sequenciamento genético de alto rendimento e sequenciamento completo do genoma ou exoma são métodos cada vez mais usados para englobar vários genes relacionados à DDS em uma única análise. O aconselhamento genético deve ser oferecido aos pais antes do exame.[8]
Exames por imagem
Uma ultrassonografia abdominal e pélvica é realizada para determinar se há presença de útero (estrutura mülleriana). Em um lactente, estrogênios maternos aumentam a capacidade de se visualizar o útero à ultrassonografia nas primeiras semanas de vida. Ovários e tubas uterinas muitas vezes não são visíveis à ultrassonografia. Uma ultrassonografia também é útil para detectar a anatomia renal, a anatomia adrenal (pois as glândulas adrenais são relativamente grandes no período neonatal), o sítio das gônadas e a morfologia.[27]
Estudos hormonais
Geralmente, essas investigações são realizadas 48 horas após o nascimento. Embora certos quadros clínicos de genitália atípica aumentem a probabilidade de alguns diagnósticos, o médico deve estar ciente de que não é possível obter um diagnóstico definitivo com base apenas no exame clínico, pois há várias apresentações em muitas DDS.
Sem gônadas palpáveis
Na ausência de gônadas palpáveis, a HAC secundária à deficiência de 21-hidroxilase deve ser excluída, pois é o diagnóstico mais provável.
Os níveis de 17-hidroxiprogesterona (17-OHP) devem ser obtidos e serão acentuadamente elevados na deficiência de 21-hidroxilase.
Os eletrólitos devem ser obtidos e monitorados rigorosamente, pois a perda de sal pode demorar alguns dias para se desenvolver, ocorrendo normalmente na segunda semana de vida em casos de deficiência à 21-hidroxilase. A medição da aldosterona e da renina ajudará a avaliar a atividade mineralocorticoide.
Se houver suspeita de HAC e os níveis de 17-OHP não estiverem acentuadamente elevados, os precursores adrenais deverão ser avaliados em detalhe em busca por causas mais raras de HAC, inclusive 11-desoxicortisol e 11-desoxicorticosterona, para descartar a deficiência de 11-beta-hidroxilase. Um perfil urinário de esteroides é útil nesse cenário.
Se a criança precisar de terapia hormonal esteroidal imediata, amostras adequadas de urina e soro deverão ser coletadas antes do início do tratamento e guardadas para análise posterior.[8]
Gônadas palpáveis
Testosterona e di-hidrotestosterona (DHT): um alto nível de testosterona em relação à DHT sugere uma deficiência de 5-alfa redutase. Um nível baixo de testosterona em relação à androstenediona sugere uma deficiência de 17 beta-hidroxiesteroide-desidrogenase.
Hormônio luteinizante (LH) e hormônio folículo-estimulante (FSH): ajudam a avaliar o eixo hipotálamo-hipofisário-gonadal. Baixos níveis na primeira semana de vida podem não necessariamente indicar hipogonadismo hipogonadotrófico; no entanto, uma avaliação aos 2 a 4 meses de vida pode ser mais reveladora quando há um aumento fisiológico nos níveis de LH e FSH.
Teste de estimulação do hormônio adrenocorticotrófico (ACTH): esse teste é usado em determinados casos para investigar anormalidades na síntese de glicocorticoides em algumas formas de DDS.
Teste de estimulação da gonadotrofina coriônica humana (hCG): esse teste avalia a capacidade das células de Leydig dos testículos de responder à hCG (um análogo do receptor do LH) e de produzir testosterona. O índice de testosterona em relação à DHT após a estimulação por hCG é usado para apurar a presença de deficiência de 5-alfa redutase. O teste de estimulação da hCG também é capaz de identificar um bloqueio da via biossintética da testosterona, como a deficiência de 17 beta-hidroxiesteroide-desidrogenase, em que a relação da testosterona para a androstenediona é baixa.
Hormônio antimülleriano (HAM) (antes chamado de substância inibidora mülleriana): pode ser obtido para avaliar a função testicular (célula de Sertoli) em um bebê com suspeita de DDS 46,XY e DDS cromossômico. Os níveis de HAM dependem da idade e do sexo.
O uso deste conteúdo está sujeito ao nosso aviso legal