Etiologia
A hiperplasia adrenal congênita (HAC), de cada variedade, é uma doença autossômica recessiva. O gene que codifica a enzima 21-hidroxilase, o CYP21A2 (família 21 do citocromo P450, subfamília A, membro 2), é mapeado no braço curto do cromossomo 6 (6p21.3). Mais de 200 mutações foram descritas, incluindo mutações pontuais, pequenas deleções, pequenas inserções e rearranjos complexos do gene.[2] Aproximadamente 95% a 98% das mutações que causam deficiência de 21-hidroxilase foram identificadas por meio de estudos de genética molecular de rearranjos gênicos e de microarranjos (arrays) de mutações pontuais.[7] A genética do gene CYP21A2 é complexa, em grande parte devido à existência de um pseudogene homólogo inativo, o CYP21AP, que hospeda muitas variações patogênicas. Os eventos de recombinação, chamados de conversões gênicas, são operacionais e levam à incorporação de mutações pontuais, deleções e duplicações de genes, bem como ao desenvolvimento de genes quiméricos. A análise genética dos pais pode precisar ser realizada em certos casos para identificar se existem duas variantes no mesmo alelo ou em alelos diferentes.[8]
Formas hormonal e clinicamente definidas de HAC deficiente em 21-hidroxilase estão associadas a genótipos distintos. [Figure caption and citation for the preceding image starts]: Mapa genético de dois homólogos: CYP21A2 (o gene ativo) e CYP21A1P (o pseudogene). As bases não correspondentes são menores que 90 em uma distância de 5.1 kb de ácido desoxirribonucleico (DNA). Os números indicam as alterações nas bases do pseudogene frequentemente identificadas nos genes CYP21A2 mutantes responsáveis pela deficiência de 21-hidroxilase (21-OHD). As mutações associadas ao NC21OHD estão indicadas com quadrados negros (exons 1, 7, 8 e 10)Imagem reproduzida com permissão de New M. Extensive personal experience: prenatal diagnosis of congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab. 2001:86;5651-5657. Copyright 2001, The Endocrine Society [Citation ends].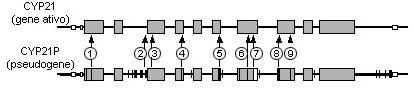 Deleções genéticas, grandes conversões genéticas e mutações que eliminam totalmente a atividade da 21-hidroxilase estão associadas à HAC clássica e são chamadas de mutações "graves". Certas mutações de sentido incorreto, como a p.Val281Leu (V281L), reduzem a atividade enzimática para aproximadamente 20%, estão associadas à HAC não clássica e são chamadas de mutações "leves".
Deleções genéticas, grandes conversões genéticas e mutações que eliminam totalmente a atividade da 21-hidroxilase estão associadas à HAC clássica e são chamadas de mutações "graves". Certas mutações de sentido incorreto, como a p.Val281Leu (V281L), reduzem a atividade enzimática para aproximadamente 20%, estão associadas à HAC não clássica e são chamadas de mutações "leves".
A forma clássica da doença é observada nos pacientes portadores de duas mutações graves. O fenótipo não clássico é causado por um genótipo leve/leve ou grave/leve, como se espera em uma doença autossômica recessiva. Entretanto, nem sempre é possível predizer com acurácia o fenótipo com base no genótipo. Estudos relatam uma concordância genótipo-fenótipo variável, de 50% a mais de 90%.[9][10]
Fisiopatologia
A produção de cortisol ocorre na zona fasciculada do córtex adrenal através de 5 etapas enzimáticas.[Figure caption and citation for the preceding image starts]: Esquema de síntese de corticosteroides adrenaisCriado pelos autores [Citation ends].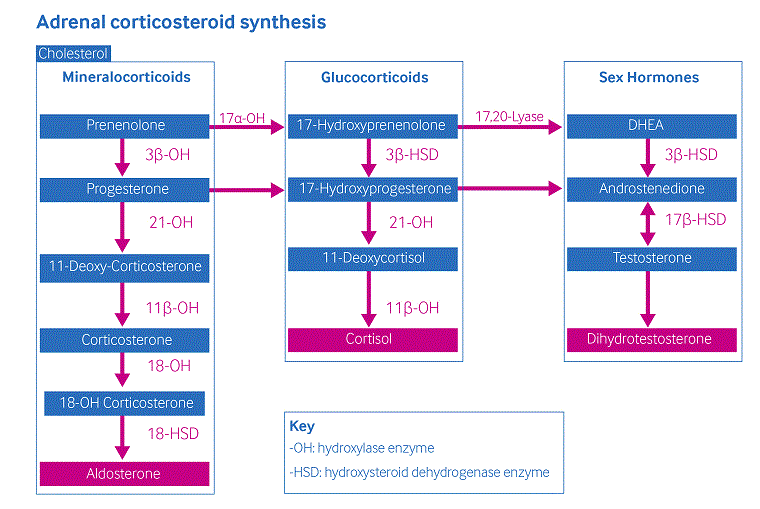
Uma produção insuficiente de cortisol devido à deficiência ou diminuição da atividade da enzima 21-hidroxilase causa produção insuficiente de cortisol, resultando em aumento da produção do hormônio liberador de corticotrofina e do hormônio adrenocorticotrófico (ACTH). Altos níveis de ACTH levam à hiperplasia adrenal e à superprodução de andrógenos adrenais (por exemplo, delta-4-androstenediona), os quais não requerem 21-hidroxilação para síntese. Sinais do excesso de androgênios são encontrados em graus variados nas formas clássica e não clássica da deficiência de 21-hidroxilase e são atribuíveis à gravidade do defeito enzimático.
Na HAC clássica, o hiperandrogenismo é significativo e resulta na virilização da genitália externa em fetos 46,XX.[2] Entretanto, o aparelho reprodutor feminino interno se desenvolve normalmente, porque o defeito esteroidogênico adrenal não afeta o desenvolvimento ovariano. O excesso de androgênios pós-parto pode se apresentar como virilização (isto é, clitoromegalia nas mulheres afetadas), crescimento rápido com maturação epifisária avançada, levando a uma baixa estatura na idade adulta, desenvolvimento prematuro de pelos pubianos e potencial para puberdade precoce central secundária à ativação periférica do eixo hipotálamo-hipófise-gonadal (HPG). O excesso de androgênios adrenais pode suprimir as gonadotrofinas hipofisárias e, assim, prejudicar o crescimento e a função gonadais.[2]
Geralmente ocorre disfunção gonadal, pois o excesso de androgênios adrenais suprime as gonadotrofinas hipofisárias e, portanto, prejudica a função e o crescimento gonadais.[2]
Quando a perda funcional da 21-hidroxilase é grave, a secreção de aldosterona adrenal é insuficiente para estimular a reabsorção de sódio pelos túbulos renais distais, resultando em perda de sal e deficiência de cortisol, além de excesso de androgênio.
Classificação
Características clínicas da HAC causada por deficiência de 21-hidroxilase[1]
A HAC devido à deficiência de 21-hidroxilase pode ser classificada como clássica ou não clássica.
A HAC clássica é tradicionalmente subdividida em duas formas, perdedora de sal e virilização simples, com base na produção relativa de mineralocorticoides. Há sobreposição entre o quadro clínico dessas duas formas, e essa subdivisão está caindo em desuso.[2]
HAC clássica: perdedora de sal
Forma mais grave da doença, caracterizada por deficiências combinadas de cortisol e aldosterona, e hiperandrogenismo adrenal grave
Aproximadamente 75% dos casos clássicos
Os bebês 46,XX podem apresentar genitália ambígua
Os bebês 46,XY (e bebês 46,XX com genitália de Prader 5) podem apresentar uma crise perdedora de sal nas primeiras 1 a 2 semanas de vida devido à ausência de percepção de uma ambiguidade. Nesses casos, o diagnóstico pode ser adiado até a fase perdedora de sal. O rastreamento neonatal visa a prevenir essa crise com risco à vida.
Pode se manifestar como órgãos genitais atípicos nas mulheres
HAC clássica: virilizante simples
O defeito enzimático é moderado, afetando a produção de cortisol, mas não a aldosterona
Aproximadamente 25% dos casos clássicos
Os bebês 46,XX podem apresentar genitália ambígua
Os bebês 46,XY podem ter um diagnóstico tardio na ausência de rastreamento neonatal adequado, mas não apresentarão perda de sal
HAC não clássica
Deficiência enzimática leve a moderada
Manifesta-se no período pós-natal com sinais de hiperandrogenismo
Meninas não apresentam órgãos genitais virilizados ao nascimento
Pode se apresentar em uma criança como desenvolvimento precoce de pelos axilares, pelos pubianos, aumento do pênis ou do clitóris, acne ou estatura alta com idade óssea avançada que pode consequentemente resultar em baixa estatura
As adolescentes e adultas do sexo feminino também podem apresentar oligomenorreia, amenorreia, ovário policístico, acne, hirsutismo, alopecia e comprometimento da fertilidade
O uso deste conteúdo está sujeito ao nosso aviso legal