O objetivo do tratamento é eliminar os efeitos perigosos da produção excessiva de catecolaminas pelo tumor. A ressecção cirúrgica é a base do tratamento.
Crise hipertensiva
Pode ocorrer uma crise hipertensiva (PA sistólica >250 mmHg) na manifestação de um feocromocitoma ou durante ressecção cirúrgica do tumor se o paciente não tiver recebido terapia medicamentosa pré-operatória adequada.
O tratamento inclui bloqueio alfa imediato com um bloqueador alfa-1 (por exemplo, terazosina, doxazosina ou prazosina) ou com o alfabloqueador não seletivo, fenoxibenzamina. Agentes intravenosos (nitroprussiato, fentolamina ou nicardipino) são de ação curta e ajustáveis e podem ser usados como primeira linha.[63]Nazari MA, Hasan R, Haigney M, et al. Catecholamine-induced hypertensive crises: current insights and management. Lancet Diabetes Endocrinol. 2023 Dec;11(12):942-54.
http://www.ncbi.nlm.nih.gov/pubmed/37944546?tool=bestpractice.com
Nitroprussiato, fentolamina ou nicardipino podem ser adicionados, conforme a necessidade, ao bloqueador alfa-1 oral no manejo inicial da crise hipertensiva.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
O quadro clínico informará as decisões relativas à prescrição. Consulte um especialista para decidir sobre o esquema mais adequado.
Bloqueio alfa pré-tratamento
O primeiro passo do tratamento clínico é bloquear os efeitos causados pelo excesso de catecolaminas, controlando a hipertensão e expandindo o volume intravascular.[1]Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019 Aug 8;381(6):552-65.
http://www.ncbi.nlm.nih.gov/pubmed/31390501?tool=bestpractice.com
Isso é alcançado ao estabelecer primeiro o bloqueio alfa adequado.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Bloqueadores alfa 1 (por exemplo, terazosina, doxazosina ou prazosina), ou o alfabloqueador não seletivo fenoxibenzamina, são recomendados para o bloqueio inicial pré-tratamento.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Esses medicamentos reduzem a PA, diminuindo a resistência vascular periférica.[64]Nicholson JP Jr, Vaughn ED Jr, Pickering TG, et al. Pheochromocytoma and prazosin. Ann Int Med. 1983 Oct;99(4):477-9.
http://www.ncbi.nlm.nih.gov/pubmed/6625381?tool=bestpractice.com
Bloqueadores alfa-1 têm uma duração de ação mais curta que a fenoxibenzamina, o que os torna particularmente úteis no período perioperatório. A dose do bloqueador alfa-1 pode ser rapidamente ajustada, evitando a hipotensão pós-operatória. Esses medicamentos não aumentam a liberação de noradrenalina e, portanto, não causam taquicardia reflexa.
Hidratação e uma dieta rica em sal (>5 g/dia) são prescritas por 7-14 dias (ou até que o paciente fique estável) para compensar os efeitos da contração do volume sanguíneo induzida pelas catecolaminas, que é associada ao bloqueio alfa.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Considere adicionar um bloqueador dos canais de cálcio ou metirosina
Bloqueadores dos canais de cálcio di-hidropiridínicos podem suplementar o bloqueio alfa, caso seja necessário o controle adicional da pressão arterial.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
A monoterapia com bloqueadores dos canais de cálcio não é recomendada, mas pode ser uma opção se o paciente não tolerar o bloqueio alfa.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
Nifedipino e anlodipino são os bloqueadores dos canais de cálcio di-hidropiridínicos mais comumente recomendados para controle perioperatório da PA nos casos de feocromocitoma.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
A metirosina pode ser usada em conjunto com o bloqueio alfa para estabilizar a pressão arterial. A metirosina, um inibidor da tirosina hidroxilase, inibe a síntese de catecolamina. Em pacientes com feocromocitomas, a metirosina pode reduzir de 35% a 80% a biossíntese das catecolaminas.[65]National Center for Biotechnology Information. PubChem Database. Metyrosine. Nov 2019 [internet publication].
https://pubchem.ncbi.nlm.nih.gov/compound/Metyrosine
Isso é particularmente útil em pacientes com níveis muito altos de catecolaminas circulantes, o que pode ser citotóxico para as células miocárdicas.
A metirosina também tem um papel em pacientes nos quais a manipulação ou a destruição de tumores será acentuada, como pacientes com doença metastática que recebem quimioterapia.[66]Tada K, Okuda T, Yamashita K. Three cases of malignant pheochromocytoma treated with cyclophosphamide, vincristine and dacarbazine combination chemotherapty and alpha-methyl-p-tyrosine to control hypercatecholaminemia. Horm Res. 1998;49(6):295-7.
http://www.ncbi.nlm.nih.gov/pubmed/9623522?tool=bestpractice.com
Deve ser iniciada 2 semanas antes da cirurgia. Também pode ser usada quando a cirurgia é contraindicada.
Bloqueio beta após bloqueio alfa
Após o bloqueio alfa adequado, que pode levar 3-4 dias de terapia, é possível estabelecer o bloqueio beta para prevenir e tratar a arritmia cardíaca e para manejo da hipertensão.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
Se apenas o bloqueio beta for utilizado, isso poderá desencadear uma crise hipertensiva em decorrência da estimulação alfa-adrenérgica sem oposição.[3]Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014 Jan-Feb;38(1):7-41.
https://www.cpcancer.com/article/S0147-0272(14)00002-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/24636754?tool=bestpractice.com
Os bloqueios alfa e beta-adrenérgicos combinados são usados com frequência para controlar a PA e prevenir a crise hipertensiva.
Feocromocitoma benigno
A excisão cirúrgica da glândula adrenal inteira permanece a base do tratamento para feocromocitomas benignos, e é curativa em mais de 85% dos casos.[67]Cryer PE. Diseases of the sympathochromaffin system. In: Felig P, Frohman LA, eds. Endocrinology and metabolism, 4th ed. New York: McGraw-Hill; 2001:525-51.[68]Fung MM, Viveros OH, O'Connor DT. Diseases of the adrenal medulla. Acta Physiol (Oxf). 2008 Feb;192(2):325-35.
http://www.ncbi.nlm.nih.gov/pubmed/18021328?tool=bestpractice.com
O tratamento de primeira escolha para feocromocitoma ≤6 cm é a adrenalectomia laparoscópica.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
A doença bilateral pode ser tratada com êxito com cirurgia preservadora da glândula adrenal.[69]Petri BJ, van Eijck CH, de Herder WW, et al. Phaeochromocytomas and sympathetic paragangliomas. Br J Surg. 2009 Dec;96(12):1381-92.
https://onlinelibrary.wiley.com/doi/full/10.1002/bjs.6821
http://www.ncbi.nlm.nih.gov/pubmed/19918850?tool=bestpractice.com
Deve-se também considerar adrenalectomia parcial em vez da total como tratamento de primeira linha para tumores adrenais pequenos.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[70]Kaye DR, Storey BB, Pacak K, et al. Partial adrenalectomy: underused first line therapy for small adrenal tumors. J Urol. 2010 Jul;184(1):18-25.
http://www.ncbi.nlm.nih.gov/pubmed/20546805?tool=bestpractice.com
A remoção é um procedimento de alto risco, que deve ser realizado por cirurgião e anestesista experientes. A excisão completa é essencial para evitar a disseminação do tumor e prevenir outros efeitos danosos da hipercatecolaminemia. As taxas de complicações cirúrgicas são baixas; foram relatadas taxas de mortalidade de cerca de 2% a 3% e taxas de morbidade de aproximadamente 20%.[71]Kinney MA, Narr BJ, Warner MA. Perioperative management of pheochromocytoma. J Cardiothorac Vasc Anesth. 2002 Jun;16(3):359-69.
http://www.ncbi.nlm.nih.gov/pubmed/12073213?tool=bestpractice.com
A adrenalectomia aberta pode ser necessária para prevenir ruptura se o tumor for grande (>6 cm), para reduzir a recorrência local de feocromocitomas invasivos, ou suspeita de malignidade.[2]Lenders JW, Duh QY, Eisenhofer G, et al; Endocrine Society. Pheochromocytoma and paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42.
https://academic.oup.com/jcem/article/99/6/1915/2537399
http://www.ncbi.nlm.nih.gov/pubmed/24893135?tool=bestpractice.com
[72]Plouin PF, Duclos JM, Soppelsa F, et al. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab. 2001 Apr;86(4):1480-6.
https://academic.oup.com/jcem/article/86/4/1480/2848250
http://www.ncbi.nlm.nih.gov/pubmed/11297571?tool=bestpractice.com
[73]Pacak KL, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. Nat Clin Pract Endocrinol Metab. 2007 Feb;3(2):92-102.
http://www.ncbi.nlm.nih.gov/pubmed/17237836?tool=bestpractice.com
Paragangliomas são tumores extra-adrenais. Eles requerem abordagens cirúrgicas especializadas, dependendo dos vários locais de origem.[69]Petri BJ, van Eijck CH, de Herder WW, et al. Phaeochromocytomas and sympathetic paragangliomas. Br J Surg. 2009 Dec;96(12):1381-92.
https://onlinelibrary.wiley.com/doi/full/10.1002/bjs.6821
http://www.ncbi.nlm.nih.gov/pubmed/19918850?tool=bestpractice.com
Risco cirúrgico
Os pacientes com maior risco de complicações são os que apresentam hipertensão pré-operatória grave, tumores com secreção elevada de catecolaminas e aqueles submetidos a repetidas intervenções cirúrgicas.[72]Plouin PF, Duclos JM, Soppelsa F, et al. Factors associated with perioperative morbidity and mortality in patients with pheochromocytoma: analysis of 165 operations at a single center. J Clin Endocrinol Metab. 2001 Apr;86(4):1480-6.
https://academic.oup.com/jcem/article/86/4/1480/2848250
http://www.ncbi.nlm.nih.gov/pubmed/11297571?tool=bestpractice.com
Muitos dos riscos associados à cirurgia são atribuídos ao risco de evolução de uma liberação maciça e descontrolada de catecolaminas decorrente da manipulação do tumor durante a ressecção. As complicações são menos prováveis com otimização clínica e a prática atual de controle rigoroso da PA perioperatória e expansão do volume plasmático.
No pós-operatório, a diminuição súbita dos níveis de catecolaminas pode ocasionar hipotensão e hipoglicemia. Pode haver hipoglicemia no pós-operatório, uma vez que supressão da secreção de insulina pelas catecolaminas deixa de existir. O tratamento envolve reposição de glicose intravenosa.[74]Akiba M, Kodama T, Ito Y, et al. Hypoglycemia induced by excessive rebound secretion of insulin after removal of pheochromocytoma. World J Surg. 1990 May-Jun;14(3):317-24.
http://www.ncbi.nlm.nih.gov/pubmed/2195784?tool=bestpractice.com
Após remoção do feocromocitoma, a secreção de catecolaminas geralmente volta ao normal em 1 semana.[Figure caption and citation for the preceding image starts]: Adrenalectomia laparoscópica esquerda: (A) exame macroscópico, tumor de 6 cm; (B) exame microscópico: neoplasia da medula adrenal com citoplasma eosinofílico de células grandes com cromogranina A granular fina positiva; (C) núcleos redondos e ovais e células sustentaculares S100+; (D) feocromocitomaAlface MM et al. BMJ Case Rep. 4 ago 2015;2015:bcr2015211184; usado com permissão [Citation ends].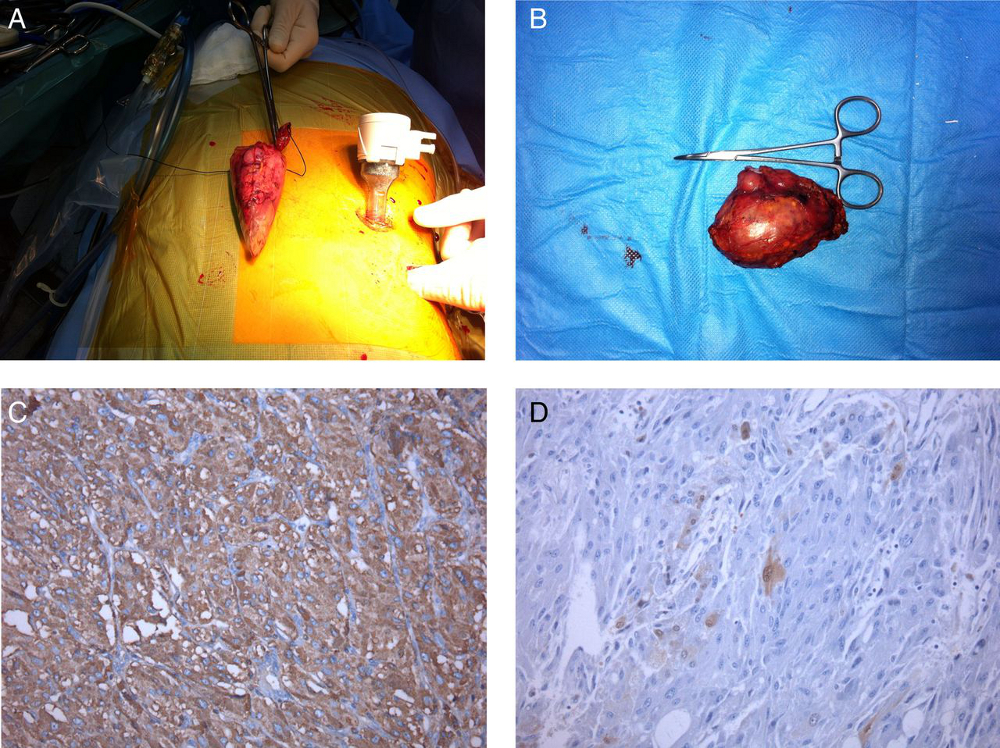
Feocromocitoma metastático
Os feocromocitomas malignos ou metastáticos representam 10% de todos os tumores secretores de catecolaminas. O diagnóstico baseia-se na invasão local dos tecidos adjacentes ou em metástases à distância.
Como na doença benigna, a remoção/citorredução cirúrgica para melhorar os sintomas é a terapia primária; no entanto, o procedimento pode não ser viável se o tumor apresentar disseminação local extensa ou metastática.[75]Adjallé R, Plouin PF, Pacak K, et al. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009 Sep;41(9):687-96.
http://www.ncbi.nlm.nih.gov/pubmed/19672813?tool=bestpractice.com
A hipertensão pode ser controlada com uma combinação de bloqueio alfa e beta-adrenérgico, além de bloqueadores dos canais de cálcio, se necessário.
A quimioterapia é administrada a todos os pacientes com doença metastática após cirurgia.[60]Kunz PL, Reidy-Lagunes D, Anthony LB, et al; North American Neuroendocrine Tumor Society. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013 May;42(4):557-77.
https://journals.lww.com/pancreasjournal/fulltext/2013/05000/Consensus_Guidelines_for_the_Management_and.2.aspx
http://www.ncbi.nlm.nih.gov/pubmed/23591432?tool=bestpractice.com
Candidatos não cirúrgico com feocromocitoma metastático também podem receber quimioterapia. Geralmente, os esquemas de quimioterapia consistem em ciclofosfamida, vincristina e dacarbazina.[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
[60]Kunz PL, Reidy-Lagunes D, Anthony LB, et al; North American Neuroendocrine Tumor Society. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013 May;42(4):557-77.
https://journals.lww.com/pancreasjournal/fulltext/2013/05000/Consensus_Guidelines_for_the_Management_and.2.aspx
http://www.ncbi.nlm.nih.gov/pubmed/23591432?tool=bestpractice.com
[76]Niemeijer ND, Alblas G, van Hulsteijn LT, et al. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014 Nov;81(5):642-51.
https://www.doi.org/10.1111/cen.12542
http://www.ncbi.nlm.nih.gov/pubmed/25041164?tool=bestpractice.com
Temozolomida, um medicamento alquilante e uma alternativa à dacarbazina, pode ser usada como monoterapia ou em combinação com outros medicamentos antineoplásicos em pacientes com feocromocitomas malignos e mutações de SDHB.[77]Tena I, Gupta G, Tajahuerce M, et al. Successful second-line metronomic temozolomide in metastatic paraganglioma: case reports and review of the literature. Clin Med Insights Oncol. 2018;12:1179554918763367.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5922490
http://www.ncbi.nlm.nih.gov/pubmed/29720885?tool=bestpractice.com
[78]Tong A, Li M, Cui Y, et al. Temozolomide is a potential therapeutic tool for patients with metastatic pheochromocytoma/paraganglioma-case report and review of the literature. Front Endocrinol (Lausanne). 2020;11:61.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7040234
http://www.ncbi.nlm.nih.gov/pubmed/32132978?tool=bestpractice.com
O radiofarmacêutico iobenguano I-131 MIBG (também conhecido como metaiodobenzilguanidina [MIBG] I-131) poderá ser considerado se o resultado da cintilografia com I-123 MIBG for positivo.
Metástases ósseas dolorosas podem ser tratadas com radioterapia por feixe externo.[79]Breen W, Bancos I, Young WF Jr, et al. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol. 2017 Nov 22;3(1):25-9.
https://www.doi.org/10.1016/j.adro.2017.11.002
http://www.ncbi.nlm.nih.gov/pubmed/29556576?tool=bestpractice.com
Ablação por radiofrequência das metástases hepáticas e ósseas também pode ser eficaz.[60]Kunz PL, Reidy-Lagunes D, Anthony LB, et al; North American Neuroendocrine Tumor Society. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013 May;42(4):557-77.
https://journals.lww.com/pancreasjournal/fulltext/2013/05000/Consensus_Guidelines_for_the_Management_and.2.aspx
http://www.ncbi.nlm.nih.gov/pubmed/23591432?tool=bestpractice.com
[80]Pacak K, Fojo T, Goldstein DS, et al. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001 Apr 18;93(8):648-9.
https://academic.oup.com/jnci/article/93/8/648/2906558
http://www.ncbi.nlm.nih.gov/pubmed/11309443?tool=bestpractice.com
Tumor irressecável
Os pacientes com tumores irressecáveis têm:
tumor maligno com disseminação local extensa ou metastática, que não pode ser removido por cirurgia, ou
tumor benigno que não seja candidato à cirurgia por outras razões clínicas (por exemplo, um paciente que tenha alto risco cirúrgico devido à insuficiência cardíaca).
O controle de longo prazo da PA com tumores irressecáveis pode ser obtido com bloqueio alfa e beta, além de bloqueadores dos canais de cálcio, se necessário.
Pacientes sintomáticos com tumores irressecáveis vêm sendo tratados com esquemas de quimioterapia (geralmente consistindo em CVD) e/ou iobenguano I-131 (também conhecido como MIBG I-131).[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
[81]Pang Y, Liu Y, Pacak K, et al. Pheochromocytomas and paragangliomas: from genetic diversity to targeted therapies. Cancers (Basel). 2019 Mar 28;11(4).
http://www.ncbi.nlm.nih.gov/pubmed/30925729?tool=bestpractice.com
No entanto, é menos provável que o tratamento baseado em iobenguano I-131 alcance a resposta completa.[82]van Hulsteijn LT, Niemeijer ND, Dekkers OM, et al. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014 Apr;80(4):487-501.
https://www.doi.org/10.1111/cen.12341
http://www.ncbi.nlm.nih.gov/pubmed/24118038?tool=bestpractice.com
Em casos de feocromocitomas malignos refratários à radioterapia e quimioterapia, sugere-se a inscrição em um ensaio clínico, e outras opções de tratamento, como inibidores de tirosina quinase (por exemplo, sunitinibe), terapia com radionuclídeo para receptor de peptídeo (lutécio Lu 177 dotatate, também conhecido como 177Lu-DOTATATE), imunoterapia (por exemplo, pembrolizumabe) e inibidores do fator 2 alfa induzível por hipóxia (por exemplo, belzutifan) devem ser considerados caso a caso.[27]Fishbein L, Del Rivero J, Else T, et al. The North American Neuroendocrine Tumor Society consensus guidelines for surveillance and management of metastatic and/or unresectable pheochromocytoma and paraganglioma. Pancreas. 2021 Apr 1;50(4):469-93.
https://nanets.net/images/2021/2021_NANETS_Consensus_Guidelines_for_Surveillance_and_Management_of_Metastatic_and_or_Unresectable_Pheochromocytoma_and_Paraganglioma.pdf
http://www.ncbi.nlm.nih.gov/pubmed/33939658?tool=bestpractice.com
[43]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: neuroendocrine and adrenal tumors [internet publication].
https://www.nccn.org/guidelines/category_1
[83]Nölting S, Bechmann N, Taieb D, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. 2022 Mar 9;43(2):199-239.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8905338
http://www.ncbi.nlm.nih.gov/pubmed/34147030?tool=bestpractice.com
[84]Jimenez C, Xu G, Varghese J, et al. New directions in treatment of metastatic or advanced pheochromocytomas and sympathetic paragangliomas: an American, contemporary, pragmatic approach. Curr Oncol Rep. 2022 Jan;24(1):89-98.
http://www.ncbi.nlm.nih.gov/pubmed/35061191?tool=bestpractice.com
Ficou comprovado que a radioterapia por feixe externo (EBRT) em doses acima de 40 Gy oferece controle do tumor local e alívio dos sintomas tanto em metástases nos tecidos moles como em metástases ósseas dolorosas.[79]Breen W, Bancos I, Young WF Jr, et al. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol. 2017 Nov 22;3(1):25-9.
https://www.doi.org/10.1016/j.adro.2017.11.002
http://www.ncbi.nlm.nih.gov/pubmed/29556576?tool=bestpractice.com
[85]Fishbein L, Bonner L, Torigian DA, et al. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Horm Metab Res. 2012 May;44(5):405-10.
http://www.ncbi.nlm.nih.gov/pubmed/22566196?tool=bestpractice.com
Paciente com feocromocitoma hereditário
Há evidências que favorecem a adrenalectomia com preservação cortical em pacientes com feocromocitomas hereditários.[69]Petri BJ, van Eijck CH, de Herder WW, et al. Phaeochromocytomas and sympathetic paragangliomas. Br J Surg. 2009 Dec;96(12):1381-92.
https://onlinelibrary.wiley.com/doi/full/10.1002/bjs.6821
http://www.ncbi.nlm.nih.gov/pubmed/19918850?tool=bestpractice.com
Esse método pode evitar a necessidade de corticoterapia vitalícia. No entanto, é necessário monitorar os pacientes no pós-operatório em relação à recorrência local.[86]Yip L, Lee JE, Shapiro SE. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004 Apr;198(4):525-34.
http://www.ncbi.nlm.nih.gov/pubmed/15051000?tool=bestpractice.com