O diagnóstico de síndrome de Noonan (SN) é tipicamente realizado clinicamente. A SN é um distúrbio clinicamente heterogêneo.
Uma história abrangente da gestação, familiar e individual, seguida por um exame físico minucioso com atenção especial às características da cabeça e da face, pescoço e tórax, é geralmente diagnóstica. Se houver suspeita de SN, o paciente deve ser investigado para as anormalidades de coagulação e cardíacas, que são comuns.
O teste genético pode ser necessário quando o diagnóstico é equívoco ou por razões familiares (por exemplo, o diagnóstico dos pais, a determinação do risco de recorrência ou o planejamento reprodutivo).
História
História gestacional e perinatal
História familiar
Alimentação
Dificuldades de alimentação ocorrem em aproximadamente 77% dos lactentes. Elas incluem dificuldade de sucção com tempo de alimentação prolongado, alimentação lenta com vômitos recorrentes ou problemas graves de alimentação exigindo alimentação por tubo durante 2 semanas ou mais.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
[31]Shah N, Rodriguez M, St Louis D, et al. Feeding difficulties and foregut dysmotility in Noonan's syndrome. Arch Dis Child. 1999 Jul;81(1):28-31.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1717976
http://www.ncbi.nlm.nih.gov/pubmed/10373129?tool=bestpractice.com
Dificuldades de alimentação estão geralmente relacionadas com hipotonia (baixo tônus muscular) e falta de coordenação da musculatura oral. No entanto, a motilidade imatura do intestino e o desenvolvimento motor gastrointestinal tardio foram documentados em alguns casos.[31]Shah N, Rodriguez M, St Louis D, et al. Feeding difficulties and foregut dysmotility in Noonan's syndrome. Arch Dis Child. 1999 Jul;81(1):28-31.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1717976
http://www.ncbi.nlm.nih.gov/pubmed/10373129?tool=bestpractice.com
O retardo do crescimento pôndero-estatural pode ocorrer em até 40% dos neonatos. Contudo, isso é autolimitado, e geralmente se resolve até os 18 meses de idade.[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
Crescimento
A história do crescimento é importante. Embora a altura ao nascer seja geralmente normal, o crescimento na infância tende a seguir o terceiro percentil, com um surto de crescimento puberal atenuado ou ausente.[32]Witt DR, Keena BA, Hall JG, et al. Growth curves for height in Noonan syndrome. Clin Genet. 1986 Sep;30(3):150-3.
http://www.ncbi.nlm.nih.gov/pubmed/3780030?tool=bestpractice.com
Puberdade
O atraso puberal é relativamente mais comum em meninos que em meninas.[33]Elsawi MM, Pryor JP, Klufio G, et al. Genital tract function in men with Noonan syndrome. J Med Genet. 1994 Jun;31(6):468-70.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049925
http://www.ncbi.nlm.nih.gov/pubmed/7915331?tool=bestpractice.com
É necessário perguntar sobre criptorquidia em meninos no nascimento.
Desenvolvimento
Comprometimento cognitivo leve é observado em um subgrupo de indivíduos com SN. O quociente de inteligência (QI) varia tipicamente de 64 a 127.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[34]Allanson JE. Noonan syndrome. J Med Genet. 1987 Jan;24(1):9-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049850
http://www.ncbi.nlm.nih.gov/pubmed/3543368?tool=bestpractice.com
Cerca de 50% dos pacientes apresentam falta de coordenação leve a moderada e outros problemas de coordenação.
Há uma probabilidade de 25% de alguma dificuldade de aprendizagem com problemas visuoconstrutivos específicos, discrepância no desempenho verbal e atraso/comprometimento da linguagem.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[35]Lee DA, Portnoy S, Hill P, et al. Psychological profile of children with Noonan syndrome. Dev Med Child Neurol. 2005 Jan;47(1):35-8.
http://www.ncbi.nlm.nih.gov/pubmed/15686287?tool=bestpractice.com
[36]Sarimski K. Developmental and behavioural phenotype in Noonan syndrome? Genet Couns. 2000;11(4):383-90.
http://www.ncbi.nlm.nih.gov/pubmed/11140417?tool=bestpractice.com
[37]Pierpont EI, Weismer SE, Roberts AE, et al. The language phenotype of children and adolescents with Noonan syndrome. J Speech Lang Hear Res. 2010 Aug;53(4):917-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3086511
http://www.ncbi.nlm.nih.gov/pubmed/20543023?tool=bestpractice.com
As crianças com SN têm um comprometimento específico no processamento global da informação visuoespacial.[38]Alfieri P, Cesarini L, De Rose P, et al. Visual processing in Noonan syndrome: dorsal and ventral stream sensitivity. Am J Med Genet A. 2011 Oct;155A(10):2459-64.
http://www.ncbi.nlm.nih.gov/pubmed/21910245?tool=bestpractice.com
Adultos com SN podem ter dificuldades específicas no processamento de informações, mas isso raramente tem um impacto na cognição.[39]Wingbermühle E, Roelofs RL, van der Burgt I, et al. Cognitive functioning of adults with Noonan syndrome: a case-control study. Genes Brain Behav. 2012 Oct;11(7):785-93.
http://onlinelibrary.wiley.com/doi/10.1111/j.1601-183X.2012.00821.x/full
http://www.ncbi.nlm.nih.gov/pubmed/22783933?tool=bestpractice.com
Os pontos fortes incluem compreensão verbal, raciocínio abstrato e consciência e julgamento social.[40]Singer ST, Hurst D, Addiego JE Jr. Bleeding disorders in Noonan syndrome: three case reports and review of the literature. J Pediatr Hematol Oncol. 1997 Mar-Apr;19(2):130-4.
http://www.ncbi.nlm.nih.gov/pubmed/9149742?tool=bestpractice.com
Os pontos fracos incluem incapacidade de organizar a informação perceptual, falta de habilidades de planejamento e falta de conhecimento espacial.
Aumento no desenvolvimento de hematomas ou sangramento
Muitos pacientes podem relatar facilidade para desenvolver hematomas.[41]Sharland M, Patton MA, Talbot S, et al. Coagulation-factor deficiencies and abnormal bleeding in Noonan's syndrome. Lancet. 1992 Jan 4;339(8784):19-21.
http://www.ncbi.nlm.nih.gov/pubmed/1345952?tool=bestpractice.com
A tendência leve a moderada ao sangramento não é incomum.[42]van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007 Jan 14;2:4.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1781428
http://www.ncbi.nlm.nih.gov/pubmed/17222357?tool=bestpractice.com
Foi relatado que a hemorragia grave ocorre em 3% dos casos.
Exame físico
A avaliação da cabeça e da face pode ser a parte mais importante para o diagnóstico no exame físico, porque determinadas características faciais são comuns, embora mudem com a idade.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[34]Allanson JE. Noonan syndrome. J Med Genet. 1987 Jan;24(1):9-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049850
http://www.ncbi.nlm.nih.gov/pubmed/3543368?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Jovem com síndrome de Noonan, observado nas idades de 3 meses (A), 2 anos (B), 6 anos (C) e 17 anos (D)Do acervo de Judith E. Allanson [Citation ends].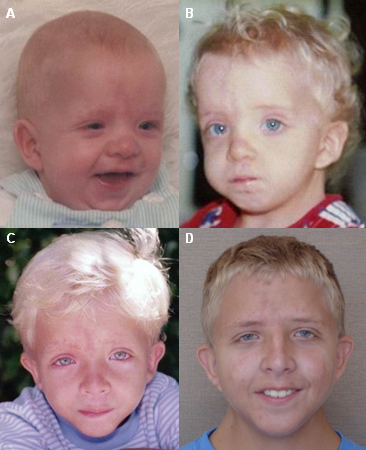 As características faciais da síndrome de Noonan são frequentemente sutis, mas talvez sejam mais acentuadas em neonatos e adolescentes. Essas características são difíceis de reconhecer em adultos, e muitos destes são diagnosticados somente após o nascimento de um filho com características mais óbvias.
As características faciais da síndrome de Noonan são frequentemente sutis, mas talvez sejam mais acentuadas em neonatos e adolescentes. Essas características são difíceis de reconhecer em adultos, e muitos destes são diagnosticados somente após o nascimento de um filho com características mais óbvias.
Cabeça e face
O neonato tem testa alta, olhos amplamente espaçados e voltados para baixo, dobras epicânticas, raiz nasal deprimida com a ponta do nariz voltada para cima, filtro labial profundamente sulcado (depressão entre o nariz e os lábios) com picos grandes e altos do vermelhão (semelhante a um arco de cupido), palato de arco alto, queixo pequeno, orelhas de implantação baixa e anguladas posteriormente com hélices espessas e excesso de pele no pescoço com linha capilar posterior baixa.
Durante a primeira infância, a cabeça parece relativamente grande, com a testa alta e proeminente. A ptose ou as pálpebras grossas e caídas são características. O nariz é curto e largo, com uma raiz deprimida.
No final da infância, a face pode parecer grosseira, hipotônica ou miopática, pois tem traços arredondados e pouca expressão. O formato facial torna-se mais triangular com a idade, à medida que a face se alonga. O paciente pode ter sobrancelhas e cílios esparsos ou ausentes.
No adolescente e no adulto jovem, o nariz tem uma ponte estreita e proeminente e uma base ampla. O pescoço é mais longo, com alas acentuadas (pterígio do pescoço) ou um músculo trapézio proeminente.
Em idosos, o sulco nasolabial incomumente proeminente e a pele fina e transparente estão presentes.[34]Allanson JE. Noonan syndrome. J Med Genet. 1987 Jan;24(1):9-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049850
http://www.ncbi.nlm.nih.gov/pubmed/3543368?tool=bestpractice.com
O perímetro cefálico médio de adultos homens é de 56.4 cm (22.2 polegadas) e nas mulheres é de 54.9 cm (21.6 polegadas).[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
Os cabelos podem ser finos e esparsos ou enrolados, grossos e lanosos.[34]Allanson JE. Noonan syndrome. J Med Genet. 1987 Jan;24(1):9-13.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1049850
http://www.ncbi.nlm.nih.gov/pubmed/3543368?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Mulher com síndrome de Noonan, observada nas idades de 4 meses (A), 4 anos (B) e adulta (C)Do acervo de Judith E. Allanson [Citation ends].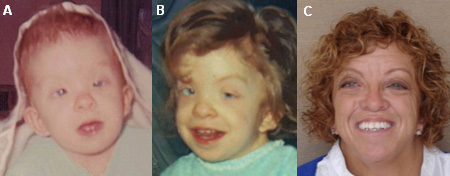
Olhos
Os achados oculares estão entre as características mais comuns da SN e são observados em até 95% dos casos.
Eles incluem estrabismo (olhos cruzados), erros de refração, ambliopia (olho vago), ptose e nistagmo (movimento ocular involuntário).
As alterações do segmento anterior, como nervos corneanos proeminentes, distrofia estromal anterior, catarata e panuveíte, foram relatadas.
Alterações do fundo são menos frequentes e incluem drusas da cabeça do nervo óptico, hipoplasia do disco óptico, colobomas e nervos mielinizados.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
[43]van Trier DC, van der Burgt I, Draaijer RW, et al. Ocular findings in Noonan syndrome: a retrospective cohort study of 105 patients. Eur J Pediatr. 2018 Aug;177(8):1293-98.
https://www.doi.org/10.1007/s00431-018-3183-1
http://www.ncbi.nlm.nih.gov/pubmed/29948256?tool=bestpractice.com
A íris geralmente é azul ou azul-esverdeada vívida, muitas vezes diferente da cor dos olhos da família, proporcionando uma excelente pista diagnóstica.
Sistema musculoesquelético
Uma deformidade do tórax específica, com pectus carinatum superiormente e pectus excavatum inferiormente, foi registrada em 70% a 95% dos casos.[Figure caption and citation for the preceding image starts]: Tórax demonstrando deformidade em funil típica, com excavatum inferior acentuado e carinatum superior sutilDo acervo de Judith E. Allanson [Citation ends].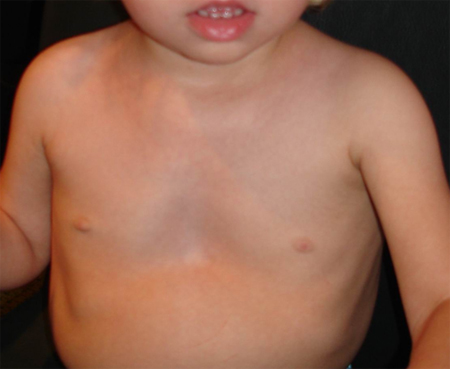
O tórax é largo, com mamilos amplamente espaçados. Durante a infância, a parte superior do tórax parece longa; os mamilos parecem ter implantação baixa e há alas axilares que persistem até a idade adulta. Os ombros podem ser arredondados, provavelmente por causa da postura hipotônica.
O cúbito valgo (um ângulo de carga elevado do antebraço) está presente em 50% dos casos, e dedos curtos com pontas rombas são observados em até um terço dos casos. A hiperextensibilidade da articulação ocorre em 30% dos casos.[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
Anomalias esqueléticas menos comuns incluem talipes equinovarus (pé torto), contraturas articulares, escoliose e sinostose radioulnar (fusão do rádio e da ulna).
Fraqueza muscular, conforme medida pela força de preensão, foi documentada.[44]Stevenson DA, Allen S, Tidyman WE, et al. Peripheral muscle weakness in RASopathies. Muscle Nerve. 2012 Sep;46(3):394-9.
http://www.ncbi.nlm.nih.gov/pubmed/22907230?tool=bestpractice.com
Sistema cardiovascular
Os defeitos cardíacos congênitos podem ocorrer em até 75% dos casos.[1]Jorge AA, Malaquias AC, Arnhold IJ, et al. Noonan syndrome and related disorders: a review of clinical features and mutations in genes of the RAS/MAPK pathway. Horm Res. 2009;71(4):185-93.
https://www.karger.com/Article/FullText/201106
http://www.ncbi.nlm.nih.gov/pubmed/19258709?tool=bestpractice.com
A anomalia mais comum é uma valva pulmonar displásica e/ou estenótica, observada em 50% a 65% das crianças afetadas.[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
Defeitos cardíacos estruturais congênitos e cardiomiopatia hipertrófica podem se apresentar no período pré-natal, período neonatal ou mais tarde na infância.
Pele
As alterações pigmentares como manchas café-com-leite, nevos pigmentados e lentigos podem ser observadas.
Os adultos podem apresentar dificuldade em deixar a barba crescer, devido à ceratose pilar atrofiante da face (pequenas depressões cicatriciais na face).[45]Pierini DO, Pierini AM. Keratosis pilaris atrophicans faciei (ulerythema ophryogenes): a cutaneous marker in the Noonan syndrome. Br J Dermatol. 1979 Apr;100(4):409-16.
http://www.ncbi.nlm.nih.gov/pubmed/454568?tool=bestpractice.com
Gestantes
Durante a gestação, as características mais comuns que sugerem o diagnóstico são polidrâmnio (excesso de líquido amniótico) e higroma cístico (lesão linfática cística).[29]Sharland M, Burch M, McKenna WM, et al. A clinical study of Noonan syndrome. Arch Dis Child. 1992 Feb;67(2):178-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1793396
http://www.ncbi.nlm.nih.gov/pubmed/1543375?tool=bestpractice.com
[46]Achiron R, Heggesh J, Grisaru D, et al. Noonan syndrome: a cryptic condition in early gestation. Am J Med Genet. 2000 May 29;92(3):159-65.
http://www.ncbi.nlm.nih.gov/pubmed/10817648?tool=bestpractice.com
[47]Benacerraf BR, Greene MF, Holmes LB. The prenatal sonographic features of Noonan's syndrome. J Ultrasound Med. 1989 Feb;8(2):59-63.
http://www.ncbi.nlm.nih.gov/pubmed/2651692?tool=bestpractice.com
[48]Houweling AC, de Mooij YM, van der Burgt I, et al. Prenatal detection of Noonan syndrome by mutation analysis of the PTPN11 and the KRAS genes. Prenat Diagn. 2010 Mar;30(3):284-6.
http://www.ncbi.nlm.nih.gov/pubmed/20112233?tool=bestpractice.com
Outras características observadas na ultrassonografia incluem edema do couro cabeludo, aumento da translucência nucal, derrame pericárdico ou pleural, ascite e/ou hidropisia (acúmulo anormal de líquido nas cavidades corporais).[46]Achiron R, Heggesh J, Grisaru D, et al. Noonan syndrome: a cryptic condition in early gestation. Am J Med Genet. 2000 May 29;92(3):159-65.
http://www.ncbi.nlm.nih.gov/pubmed/10817648?tool=bestpractice.com
[49]Bawle EV, Black V. Nonimmune hydrops fetalis in Noonan's syndrome. Am J Dis Child. 1986 Aug;140(8):758-60.
http://www.ncbi.nlm.nih.gov/pubmed/3728401?tool=bestpractice.com
Esplenomegalia
Aumento do baço foi observado e pode ser uma característica da mielodisplasia.[40]Singer ST, Hurst D, Addiego JE Jr. Bleeding disorders in Noonan syndrome: three case reports and review of the literature. J Pediatr Hematol Oncol. 1997 Mar-Apr;19(2):130-4.
http://www.ncbi.nlm.nih.gov/pubmed/9149742?tool=bestpractice.com
Malformações renais
Encontradas em até 10% dos casos, essas características incluem malformações como sistema coletor duplex, estenose ureteral distal, hipoplasia renal, agenesia renal unilateral e ectopia renal unilateral.[50]George CD, Patton MA, El Sawi M, et al. Abdominal ultrasound in Noonan syndrome: a study of 44 patients. Pediatr Radiol. 1993;23(4):316-8.
http://www.ncbi.nlm.nih.gov/pubmed/8414765?tool=bestpractice.com
Critérios diagnósticos: escores
Vários escores foram criados para ajudar o processo diagnóstico.[42]van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007 Jan 14;2:4.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1781428
http://www.ncbi.nlm.nih.gov/pubmed/17222357?tool=bestpractice.com
[51]Duncan WJ, Fowler RS, Farkas LG, et al. A comprehensive scoring system for evaluating Noonan syndrome. Am J Med Genet. 1981;10(1):37-50.
http://www.ncbi.nlm.nih.gov/pubmed/7294061?tool=bestpractice.com
O escore mais recente foi desenvolvido em 1994.[42]van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007 Jan 14;2:4.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1781428
http://www.ncbi.nlm.nih.gov/pubmed/17222357?tool=bestpractice.com
Nesse sistema, a síndrome de Noonan (SN) é definida como características faciais típicas associadas a 1 característica clínica primária ou 2 secundárias, ou características faciais sugestivas associadas a 2 características clínicas primárias ou 3 secundárias. No entanto, as características faciais típicas podem ser sutis e frequentemente requerem a avaliação de um dismorfologista experiente. O teste molecular para mutações nos genes que são causadores conhecidos da SN pode ser necessário se o diagnóstico for questionável.
Características primárias
Cardíaco: estenose pulmonar valvar e/ou eletrocardiograma (ECG) típico
Altura: <terceiro percentil
Parede torácica: pectus carinatum/excavatum
História familiar: parente de primeiro grau com diagnóstico definido
Comprometimento cognitivo, criptorquidia, displasia linfática: todos os 3 presentes.
Características secundárias
Cardíaco: outros defeitos afora os descritos como características primárias
Altura: <décimo percentil; a baixa estatura pode estar presente ao nascimento ou ter uma origem pós-parto
Tórax: tórax largo
Família: parente de primeiro grau com diagnóstico sugestivo
Comprometimento cognitivo, criptorquidia, displasia linfática: qualquer um dos 3 presente.
Investigações laboratoriais
Os relatórios documentaram vários tipos de defeitos de coagulação e diáteses hemorrágicas na SN e uma grande variedade de quadros clínicos.[52]Derbent M, Oncel Y, Tokel K, et al. Clinical and hematologic findings in Noonan syndrome patients with PTPN11 gene mutations. Am J Med Genet A. 2010 Nov;152A(11):2768-74.
http://www.ncbi.nlm.nih.gov/pubmed/20954246?tool=bestpractice.com
Se houver suspeita de síndrome de Noonan, o paciente deve fazer o rastreamento para anormalidades de coagulação. Inicialmente, um hemograma completo com contagem plaquetária e perfil de coagulação (isto é, tempo de protrombina, tempo de tromboplastina parcial ativada, tempo de sangramento) pode ser considerado.[53]Briggs BJ, Dickerman JD. Bleeding disorders in Noonan syndrome. Pediatr Blood Cancer. 2012 Feb;58(2):167-72.
http://www.ncbi.nlm.nih.gov/pubmed/22012616?tool=bestpractice.com
Homens com evidência de atraso puberal devem fazer uma avaliação da função testicular, pois a disfunção da célula de Sertoli foi descrita.[54]Marcus KA, Sweep CG, van der Burgt I, et al. Impaired Sertoli cell function in males diagnosed with Noonan syndrome. J Pediatr Endocrinol Metab. 2008 Nov;21(11):1079-84.
http://www.ncbi.nlm.nih.gov/pubmed/19189703?tool=bestpractice.com
Outras investigações
ECG e ecocardiografia
Os defeitos cardíacos congênitos estão presentes em até 75% dos casos, portanto, um exame cardiovascular completo deve ser realizado.[1]Jorge AA, Malaquias AC, Arnhold IJ, et al. Noonan syndrome and related disorders: a review of clinical features and mutations in genes of the RAS/MAPK pathway. Horm Res. 2009;71(4):185-93.
https://www.karger.com/Article/FullText/201106
http://www.ncbi.nlm.nih.gov/pubmed/19258709?tool=bestpractice.com
O ECG e a ecocardiografia devem identificar as características das anomalias cardíacas mais comuns (isto é, valva pulmonar displásica e/ou estenótica, cardiomiopatia hipertrófica, defeitos do septo e tetralogia de Fallot).[30]Shaw AC, Kalidas K, Crosby AH, et al. The natural history of Noonan syndrome: a long-term follow-up study. Arch Dis Child. 2007 Feb;92(2):128-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2083343
http://www.ncbi.nlm.nih.gov/pubmed/16990350?tool=bestpractice.com
[55]Burch M, Sharland M, Shinebourne E, et al. Cardiologic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol. 1993 Oct;22(4):1189-92.
http://www.sciencedirect.com/science/article/pii/0735109793904365?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/8409059?tool=bestpractice.com
[56]Ishizawa A, Oho S, Dodo H, et al. Cardiovascular abnormalities in Noonan syndrome: the clinical findings and treatments. Acta Paediatr Jpn. 1996 Feb;38(1):84-90.
http://www.ncbi.nlm.nih.gov/pubmed/8992869?tool=bestpractice.com
[57]Noonan JA, O'Connor W. Noonan syndrome: a clinical description emphasizing the cardiac findings. Acta Paediatr Jpn. 1996 Feb;38(1):76-83.
http://www.ncbi.nlm.nih.gov/pubmed/8992867?tool=bestpractice.com
Teste genético molecular
Pode ser necessário quando o diagnóstico é equívoco ou por razões familiares (por exemplo, o diagnóstico dos pais, a determinação do risco de recorrência ou o planejamento reprodutivo).
Estão disponíveis testes diagnósticos clínicos para os genes da via Ras/MAPK, causadores conhecidos da SN. No entanto, uma proporção dos portadores de SN não terá mutação alguma nos genes que atualmente são causadores conhecidos dessa afecção.
Mutações do PTPN11 são encontradas em 50% a 60% dos indivíduos afetados.[11]Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001 Dec;29(4):465-8.
http://www.ncbi.nlm.nih.gov/pubmed/11704759?tool=bestpractice.com
[12]Tartaglia M, Kalidas K, Shaw A, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002 Jun;70(6):1555-63.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC379142
http://www.ncbi.nlm.nih.gov/pubmed/11992261?tool=bestpractice.com
Mutações do SOS1 são encontradas em cerca de 10% a 15% dos casos.[3]Roberts AE, Araki T, Swanson KD, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007 Jan;39(1):70-4.
http://www.ncbi.nlm.nih.gov/pubmed/17143285?tool=bestpractice.com
[4]Tartaglia M, Pennacchio LA, Zhao C, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007 Jan;39(1):75-9.
http://www.ncbi.nlm.nih.gov/pubmed/17143282?tool=bestpractice.com
Mutações do RAF1 são encontradas em cerca de 5% dos casos.[5]Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007 Aug;39(8):1007-12.
http://www.ncbi.nlm.nih.gov/pubmed/17603483?tool=bestpractice.com
[6]Razzaque MA, Nishizawa T, Komoike Y, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007 Aug;39(8):1013-7.
http://www.ncbi.nlm.nih.gov/pubmed/17603482?tool=bestpractice.com
Mutações do KRAS são encontradas em cerca de 1% dos casos.[7]Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006 Mar;38(3):331-6.
http://www.ncbi.nlm.nih.gov/pubmed/16474405?tool=bestpractice.com
[16]Carta C, Pantaleoni F, Bocchinfuso G, et al. Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006 Jul;79(1):129-35.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1474118
http://www.ncbi.nlm.nih.gov/pubmed/16773572?tool=bestpractice.com
Raramente, mutações em outros genes (por exemplo, NRAS, BRAF ou MAP2K1) serão identificadas.[8]Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010 Jan;42(1):27-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3118669
http://www.ncbi.nlm.nih.gov/pubmed/19966803?tool=bestpractice.com
[9]Nyström AM, Ekvall S, Berglund E, et al. Noonan and cardio-facio-cutaneous syndromes: two clinically and genetically overlapping disorders. J Med Genet. 2008 Aug;45(8):500-6.
http://www.ncbi.nlm.nih.gov/pubmed/18456719?tool=bestpractice.com
[10]Sarkozy A, Carta C, Moretti S, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009 Apr;30(4):695-702.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4028130
http://www.ncbi.nlm.nih.gov/pubmed/19206169?tool=bestpractice.com
Os pontos de ‘hotspot’ da mutação são observados em alguns desses genes.
Alguns laboratórios sugerem testes de forma escalonada, começando com os pontos de hotspot do PTPN11 (teste de gene único em série). Outros laboratórios usam um painel multigênico que testa simultaneamente todos os genes conhecidos por causar a síndrome de Noonan.
Em gestações complicadas por higroma cístico, aumento da translucência nucal ou hidropisia, em que a análise dos cromossomos é normal, mutações do PTPN11 são encontradas em 3% a 11% dos casos, dependendo do achado na ultrassonografia.[48]Houweling AC, de Mooij YM, van der Burgt I, et al. Prenatal detection of Noonan syndrome by mutation analysis of the PTPN11 and the KRAS genes. Prenat Diagn. 2010 Mar;30(3):284-6.
http://www.ncbi.nlm.nih.gov/pubmed/20112233?tool=bestpractice.com
[58]Lee KA, Williams B, Roza K, et al. PTPN11 analysis for the prenatal diagnosis of Noonan syndrome in fetuses with abnormal ultrasound findings. Clin Genet. 2009 Feb;75(2):190-4.
http://www.ncbi.nlm.nih.gov/pubmed/18759865?tool=bestpractice.com
Ultrassonografia abdominal
Deve ser realizada se houver suspeita de aumento do baço. Esplenomegalia pode ser uma característica da mielodisplasia.[40]Singer ST, Hurst D, Addiego JE Jr. Bleeding disorders in Noonan syndrome: three case reports and review of the literature. J Pediatr Hematol Oncol. 1997 Mar-Apr;19(2):130-4.
http://www.ncbi.nlm.nih.gov/pubmed/9149742?tool=bestpractice.com
Ultrassonografia renal
Deve ser realizada se houver suspeita de malformação renal (por exemplo, como sistema coletor duplex, estenose ureteral distal, hipoplasia renal, agenesia renal unilateral ou ectopia renal unilateral); essas malformações podem estar presentes em até 10% dos casos.[50]George CD, Patton MA, El Sawi M, et al. Abdominal ultrasound in Noonan syndrome: a study of 44 patients. Pediatr Radiol. 1993;23(4):316-8.
http://www.ncbi.nlm.nih.gov/pubmed/8414765?tool=bestpractice.com