História e exame físico
Principais fatores diagnósticos
comuns
presença de fatores de risco
Muitos pacientes com retinite pigmentosa (RP) ligada ao cromossomo X ou dominante terão uma história familiar de RP. A herança autossômica recessiva pode ou não revelar uma história familiar de degeneração retiniana. Geralmente, a RP é um achado isolado, mas pode ser detectada como característica de um distúrbio genético raro quando em combinação com outros sintomas, como problemas de equilíbrio, perda auditiva e doença renal.
redução da visão periférica
Característica principal da forma mais comum de bastonetes-cones de RP. A perda do campo de visão frequentemente não é notada até que esta atinge um estágio moderado devido à sobreposição dos campos visuais de cada olho.
cegueira noturna
Geralmente um dos primeiros sintomas. Muitos pacientes relatam que desistiram de dirigir à noite devido a dificuldades em enxergar.
comprometimento da adaptação ao escuro
Os pacientes frequentemente relatam dificuldade de entrar em cinemas ou, ao contrário, descrevem problemas quando saem para um ambiente com bastante luz.
acuidade central reduzida
Pode ser observada em formas avançadas do tipo de doença mais comum, a distrofia bastonetes-cones, ou em um momento anterior, se o edema macular cistoide se desenvolver. As formas de cones-bastonetes de RP podem se manifestar com diminuição precoce na acuidade central.
atrofia do epitélio pigmentar da retina
Uma característica chave. Dependendo da mutação envolvida, pode ocorrer através da degeneração primária do epitélio pigmentar da retina ou secundária à degeneração dos fotorreceptores.
pigmentação em espículas ósseas
Uma característica chave, resultado da migração das células do epitélio pigmentar da retina para a camada de fotorreceptores da mesma, frequentemente circundando os vasos retinianos.[Figure caption and citation for the preceding image starts]: Espículas ósseasDo acervo do Oregon Retinal Degeneration Center [Citation ends].
Outros fatores diagnósticos
comuns
nervo óptico pálido em cera
Acredita-se que seja causado por gliose ao redor do nervo óptico.[Figure caption and citation for the preceding image starts]: Palidez em cera e atenuação vascularDo acervo do Oregon Retinal Degeneration Center [Citation ends].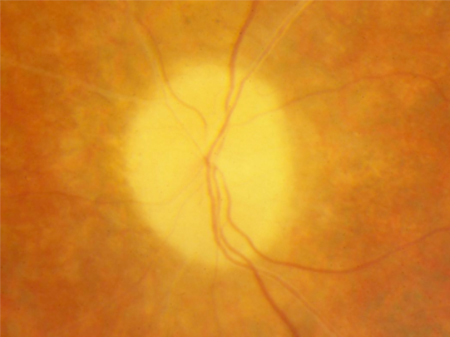
fotopsias
Flashes de luz contínuos, experimentados comumente. É importante distinguir essas fotopsias de flashes devido a descolamento da retina ou em associação com enxaquecas.
erro de refração
Em formas graves precoces de RP, como a amaurose congênita de Leber, a hiperopia predomina. Em formas de início tardio, a miopia e o astigmatismo são mais comuns.
catarata
O desenvolvimento precoce de catarata polar ou subcapsular posteriores é especialmente comum.
atenuação vascular retiniana
Acredita-se que seja secundária à atrofia retiniana. Sua presença pode ajudar a distinguir a RP da coroideremia, que é caracterizada por vasos retinianos normais e pela atrofia dos vasos coroidais.[Figure caption and citation for the preceding image starts]: Palidez em cera e atenuação vascularDo acervo do Oregon Retinal Degeneration Center [Citation ends].
edema macular cistoide
Complicação comum. A tomografia de coerência óptica é o melhor método de detecção.
células vítreas
Células vítreas leves são frequentemente observadas em um exame com lâmpada de fenda do fundo do olho. Uma inflamação mais significativa deve levar à suspeita de outras doenças que podem mimetizar a RP.
ofuscamento por luzes fortes
Pode ser um problema em casos de doença mais avançada.
Incomuns
visão das cores anormal
Pode ser observada precocemente em distrofias cones-bastonetes ou mais tarde, em distrofias bastonetes-cones. Geralmente afeta o eixo azul-amarelo, o que ajuda a distingui-la da cegueira mais comum vermelho-verde, ligada ao cromossomo X, que está presente em cerca de 10% da população masculina.
ceratocone
Mais comum em pacientes com RP.
glaucoma
Mais comum em pacientes com RP.
drusas da cabeça do nervo óptico
Anomalias congênitas e do desenvolvimento da cabeça do nervo óptico. Elas são formadas por degeneração cálcica em alguns axônios do nervo óptico. Frequentemente, são encontradas de maneira incidental, mas são mais comuns em pacientes com RP. Com o tempo, elas podem crescer e invadir a camada de fibras nervosas da retina, causando defeitos no campo de visão. Uma razão escavação/disco pequena pode indicar drusas ópticas enterradas.
retinopatia tipo Coats
Um sinal raro em certos pacientes com RP. É caracterizada por telangiectasias vasculares anormais em áreas periféricas da retina, que podem extravasar e causar exsudação da retina.
amaurose congênita de Leber
Esse é um subtipo grave de RP, e pode se manifestar na primeira infância com visão diminuída, pupilas lentas e nistagmo.[5]
Fatores de risco
Fortes
história familiar
A herança pode ser autossômica dominante, autossômica recessiva, ligada ao cromossomo X ou, raramente, mitocondrial ou digênica. Uma história familiar pode ajudar a deduzir o modo de herança. A prevalência de cada tipo varia em diferentes regiões. A maioria dos pacientes que herda mutações genéticas exibirá os sintomas da doença, mas a gravidade pode variar muito, especialmente em formas autossômicas dominantes, que podem mostrar penetrância e expressividade diversas.[19]
presença de uma síndrome associada
Geralmente, a retinite pigmentosa (RP) é um achado isolado, mas pode ser detectada como característica de algum distúrbio genético raro quando em combinação com outros sintomas, como problemas de equilíbrio, perda auditiva e doença renal. Esses distúrbios podem incluir: síndrome de Usher, de Bardet-Biedl, de Alstrom, de Joubert, de Senior-Loken, lipofuscinose ceroide neuronal, síndrome de Kearns-Sayre, doença de Bassen-Kornzweig (abetalipoproteinemia) e doença de Refsum adulta ou infantil.[20][21]
O uso deste conteúdo está sujeito ao nosso aviso legal