Etiologia
A retinite pigmentosa (RP) é causada por mutações em genes que codificam proteínas importantes na função e na sobrevivência dos fotorreceptores e do epitélio pigmentar da retina. Mutações em mais de 100 genes individuais foram apontadas como causadoras da RP. As funções desses diferentes genes são diversas, incluindo fototransdução, ciclo retinoide, integridade estrutural dos fotorreceptores, fagocitose das pontas dos segmentos externos, estrutura e função dos cílios, homeostase metabólica, splicing do RNAm e transcrição de proteínas. Uma história familiar de RP pode ajudar a revelar o padrão da herança.
A RP é, frequentemente, um achado ocular isolado. No entanto, existem algumas formas raras de RP sindrômica, que se manifestam com achados sistêmicos em associação com uma degeneração retiniana pigmentar típica. As formas mais comuns da RP sindrômica são a síndrome de Usher e a síndrome de Bardet-Biedl.[17] Nas formas sindrômicas, os genes afetados têm impacto na função dos fotorreceptores, mas, adicionalmente, exercem uma função importante em outros sistemas de órgãos, como a orelha ou os rins.
Síndromes associadas à RP
Síndrome de Usher: esta síndrome afeta a audição e a visão devido à RP e a uma orelha interna defeituosa. Muitos pacientes também apresentam problemas de equilíbrio, e crianças com esta doença podem demorar para começar a andar.
Síndrome de Bardet-Biedl: geralmente, há comprometimento intelectual e do crescimento, com características como polidactilia ou sindactilia, cardiomiopatia dilatada, insuficiência renal e fibrose hepática. O grau de comprometimento mental pode variar de um comprometimento cognitivo leve a deficiência intelectual grave.
Síndrome de Alstrom: uma doença autossômica recessiva rara. As características incluem RP (distrofia cones-bastonetes), perda auditiva sensorioneural, cardiomiopatia infantil e obesidade. Na ausência de cardiomiopatia, a doença pode não ser diagnosticada até o desenvolvimento tardio de diabetes mellitus.
Síndrome de Joubert: uma doença rara, geralmente hereditária de uma forma autossômica recessiva que resulta em malformação do vérmis cerebelar (que conecta as 2 metades do cerebelo). A ataxia é uma característica principal, mas outras manifestações incluem dificuldade de aprendizagem, tônus muscular diminuído, cistos renais, apneia do sono e dedos e pododáctilos extras.
Síndrome de Senior-Loken: um distúrbio raro, autossômico recessivo, causado por mutação em um gene envolvido na formação proteica nos cílios. A RP é acompanhada de doença renal progressiva causada pelo funcionamento defeituoso dos néfrons e pela formação de cistos medulares. A doença renal tipicamente se manifesta no primeiro ano de vida.
Lipofuscinose ceroide neuronal: uma família de raros distúrbios genéticos neurodegenerativos de depósito lipossômico. Há degeneração neurológica e motora graduais, convulsões e perda da visão devida à RP. A perda visual pode ser uma característica precoce.
Síndrome de Kearns-Sayre: uma doença mitocondrial que tipicamente se manifesta em indivíduos <20 anos de idade com perda da visão devida à RP. Outras características podem incluir oftalmoplegia, perda auditiva, ataxia e disfagia.
Doença de Bassen-Kornzweig (abetalipoproteinemia): uma doença rara, autossômica recessiva, que resulta em má absorção de gorduras, vitaminas lipossolúveis (A, D, E, K) e colesterol. Os sintomas geralmente se manifestam dentro dos primeiros meses de vida e incluem retardo do crescimento pôndero-estatural, crescimento deficiente e presença de gordura e sangue nas fezes. A RP e a ataxia podem se desenvolver durante a segunda infância.
Doença de Refsum infantil: um distúrbio metabólico que ocorre devido a um defeito na biogênese peroxissomal. Este distúrbio geralmente se manifesta na primeira infância com comprometimento da audição e da visão, tônus muscular e coordenação prejudicados, comprometimento intelectual e desenvolvimento facial anormal.
Doença de Refsum adulta: pode-se suspeitar dessa doença em crianças que, na terceira infância, apresentem RP e combinações de anosmia, perda auditiva, ataxia, arritmias cardíacas e metacarpos e metatarsos curtos. Alguns apresentam neuropatia motora ou sensorial.
Fisiopatologia
Embora a retinite pigmentosa (RP) possa ser causada por mutações de uma série de proteínas funcionalmente diferentes, a característica fisiopatológica subjacente comum é a apoptose dos cones e bastonetes fotorreceptores. Os cones são arranjados centralmente na retina, e a perda dos cones fotorreceptores comprometerá os campos de visão sob condições de luz adaptada (fotópica), podendo resultar em sensibilidade à luz, acuidade visual diminuída e perda da visão das cores. A perda de bastonetes fotorreceptores causa o comprometimento da visão e campos visuais reduzidos sob condições de pouca luz (escotópica), além de problemas de adaptação ao escuro. O mais comum, são os bastonetes fotorreceptores se degenerarem antes dos cones (distrofia bastonetes-cones), mas certos genes podem causar uma distrofia cones-bastonetes, onde os cones são os primeiros que se degeneram e depois os bastonetes. Finalmente, algumas mutações causam uma distrofia de cones isolada.
Ao longo do processo de degeneração dos fotorreceptores, o epitélio pigmentar da retina migra para dentro da camada neural da retina para formar as espículas ósseas que circundam os vasos retinianos.[18] Uma desorganização secundária da camada interna da retina também é observada.
Classificação
Classificação por herança
Autossômica dominante
Ligado ao cromossomo X
Autossômica recessiva
Simples: nenhuma história familiar, presume-se que seja recessiva, mas também pode ser uma mutação dominante "de novo"
Múltipla: autossômica recessiva com outros irmãos afetados.
Mitocondrial (rara)
Digênica (rara)
Classificação por perda funcional
Distrofia de bastonetes-cones
Distrofia de cones-bastonetes
Distrofia de cones
Classificação por idade no início
Amaurose congênita de Leber: mutações em genes que causam doença grave com início em <1 ano de idade
RP juvenil: mutações em genes que causam doença grave com início entre 1 e 5 anos de idade
RP típica: idade de início variável >5 anos de idade
Classificação por padrões de fundo do olho observados no exame do olho
Padrão clássico: começa na porção centro-periférica e se estende à periferia e, em seguida, centralmente[Figure caption and citation for the preceding image starts]: Retinite pigmentosaDo acervo do Oregon Retinal Degeneration Center [Citation ends].
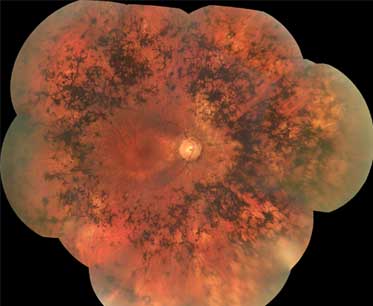
Maculopatia em "olho de boi": observada em distrofias de cones-bastonetes e de cones (rara na RP típica)[Figure caption and citation for the preceding image starts]: Aparência tipo "olho de boi" na retinite pigmentosa (RP)Do acervo do Oregon Retinal Degeneration Center [Citation ends].
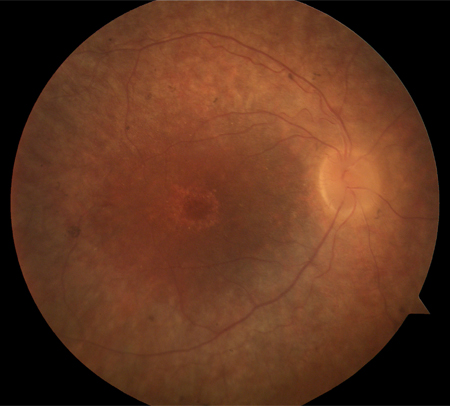
RP setorial : envolve 1 ou mais regiões da retina em ambos os olhos[Figure caption and citation for the preceding image starts]: Retinite pigmentosa (RP) setorialDo acervo do Oregon Retinal Degeneration Center [Citation ends].
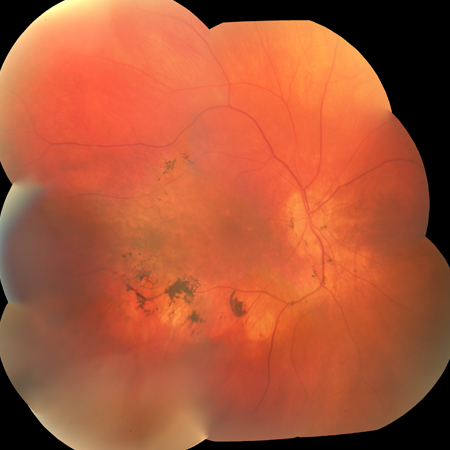
Padrão em mosaico: pode ser observado em mulheres portadoras de mutações ligadas ao cromossomo X
RP pericentral: começa pericentralmente dentro das arcadas e se estende para o centro
Concêntrica: começa na porção periférica distal e evolui centralmente
Unilateral: provavelmente não é RP, mas uma doença que mimetiza a RP
RM sem pigmento: não apresenta a pigmentação em espículas ósseas característica; pode ser observada no início da doença
RP com epitélio pigmentar da retina periarteriolar preservado: padrão clássico, mas com a retina preservada perto das arteríolas
Atrofia retinocoroidiana pigmentada paravenosa: espículas ósseas limitadas à área ao redor das veias retinianas.
O uso deste conteúdo está sujeito ao nosso aviso legal