Etiología
La glomeruloesclerosis focal y segmentaria (GEFS) se inicia por una lesión de los podocitos en los glomérulos renales. En la GEFS primaria, esta lesión es producida por factores circulantes hasta ahora no identificados que alteran la permeabilidad de los glomérulos.[22] Las moléculas candidatas propuestas incluyen la hemopexina, el factor de crecimiento endotelial vascular, la forma soluble del receptor de urocinasa (suPAR) y la citocina-1 similar a la cardiotrofina.
suPAR es una molécula candidata que ha sido ampliamente investigada. En un estudio, se observaron niveles elevados de suPAR en el plasma de dos tercios de los pacientes con GEFS.[23] Un metanálisis también demostró que los niveles de suPAR eran significativamente más altos en los pacientes con GEFS primaria en comparación con los controles.[24]
Algunas variantes de la GEFS primaria son hereditarias, y los antecedentes familiares positivos pueden sugerir una causa genética. Las mutaciones en una variedad de proteínas de los podocitos se han asociado con la GEFS.[25] La mayoría de las causas genéticas en niños siguen un patrón autosómico recesivo, siendo las mutaciones en los genes nefrina (NPHS1), podocina (NPHS2) y fosfolipasa C épsilon 1 (PLCE1) las más frecuentes.[26] Las causas autosómicas dominantes de GEFS son más frecuentes en adolescentes y adultos. Los principales genes implicados en la GEFS autosómica dominante incluyen la alfa-actinina 4 (ACTN4), la formina invertida 2 (INF2) y el canal catiónico de potencial del receptor transitorio 6 (TRPC6).[25] Las mutaciones MYO1E se han asociado con la GEFS resistente a los glucocorticoides de inicio en la infancia.[27]
En la GEFS secundaria, la lesión podocitaria se produce por virus, toxinas, cambios hemodinámicos intrarrenales o por una respuesta inadaptada a la pérdida de nefronas funcionantes. No obstante, no queda claro por qué el daño que inducido estas diversas causas se dirige específicamente a los podocitos glomerulares.
Las afecciones subyacentes que producen la GEFS secundaria incluyen:
Infección viral: el VIH es la causa más frecuente. Las causas más raras incluyen la infección persistente por parvovirus B19, citomegalovirus, hepatitis B y C y SARS-CoV-2.
Hiperfiltración glomerular por masa renal reducida: la masa renal puede verse reducida por tener un riñón único (debido a nefrectomía o agenesia renal unilateral), nefropatía por reflujo, displasia renal o nefropatía crónica del aloinjerto (en receptores de un trasplante renal).
Inducida por fármacos: heroína, interferón alfa, litio, pamidronato y sirolimús son causas conocidas.
Obesidad.
Vascular: la tromboembolia y la isquemia renal son causas poco frecuentes.
Los pacientes de raza negra tienen un riesgo mayor de desarrollar insuficiencia renal terminal debida a GEFS primaria o secundaria y tienden a desarrollar la insuficiencia renal a una edad más joven que los pacientes de raza blanca o asiática. El aumento de la propensión a la GEFS puede estar relacionado a las mutaciones en MYH9, que son más comunes en pacientes de raza negra. Estas mutaciones pueden causar GEFS primaria y exacerban la GEFS inducida por el VIH. Los polimorfismos del gen de la apolipoproteína L1 (APOL1) también se asocian con la GEFS en pacientes de raza negra.[28] Estos parecen expresarse casi exclusivamente en individuos de ascendencia africana, pero se han identificado con menos frecuencia en otras poblaciones.[29]
Fisiopatología
La lesión de los podocitos desencadena apoptosis, lo cual hace que se desprendan de la membrana basal glomerular y sean destruidos.[1] Como el número de podocitos se reduce, la membrana basal glomerular queda expuesta y se desarrollan interacciones inadecuadas entre la membrana basal glomerular y las células epiteliales parietales. A esto le sigue la proliferación de células epiteliales, endoteliales y mesangiales. La combinación de la proliferación celular y la filtración de proteínas hacia el espacio de Bowman da como resultado la deposición de colágeno. Finalmente, se colapsa el penacho capilar y se pierden las células endoteliales.[30] El ovillo glomerular se esclerosa, lo cual crea la lesión característica de la glomeruloesclerosis focal y segmentaria (GEFS). Inicialmente, la extensión de las lesiones varía entre las diferentes regiones del riñón. Sin embargo, la enfermedad finalmente avanza hasta producir esclerosis global e insuficiencia renal terminal.[Figure caption and citation for the preceding image starts]: Aspecto característico de las lesiones de la glomeruloesclerosis focal y segmentariaCreado en el BMJ Knowledge Centre [Citation ends].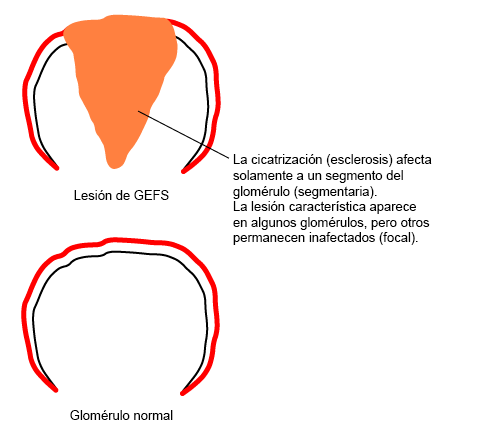
Una vez que los podocitos comienzan a morir, las proteínas se filtran a través de la membrana glomerular, lo que conduce a proteinuria e hipoalbuminemia. Al mismo tiempo, aumentan los niveles de colesterol debido a una mayor síntesis de colesterol en el hígado y a la pérdida de proteínas reguladoras de lípidos por la orina; el mecanismo de estos efectos no se comprende por completo.
La gravedad de la GEFS difiere entre GEFS primaria y secundaria. La GEFS primaria produce una lesión más grave de los podocitos y se asocia más frecuentemente con el síndrome nefrótico. La lesión producida por la GEFS secundaria es menos grave y la proteinuria con hipoalbuminemia en rango nefrótico es menos frecuente. Esta diferencia se refleja en el patrón de la lesión de los podocitos detectada por la microscopía electrónica. La GEFS produce la fusión de las prolongaciones primarias de los podocitos.[31] En la GEFS primaria, la fusión de las prolongaciones primarias es difusa y ocurre en todo el glomérulo. En la GEFS secundaria, la fusión de las prolongaciones primarias se limita principalmente a las áreas escleróticas.
Clasificación
Clasificación etiológica
GEFS primaria (idiopática)
Mediada por factores circulantes/permeables no identificados y caracterizada por borramiento difuso de procesos podocitarios y síndrome nefrótico.
GEFS secundaria
Causada por una afección subyacente adquirida.
GEFS genética
Se desarrolla en pacientes con mutaciones en genes que codifican proteínas de podocitos o de la membrana basal glomerular.
GEFS de causa indeterminada
Se caracteriza por borramiento segmentario de procesos podocitarios y proteinuria sin síndrome nefrótico, sin evidencia de causas secundarias.
Clasificación morfológica[2][3]
Variante perihiliar
Al menos 1 glomérulo tiene hialinosis perihiliar, con o sin esclerosis.
>50% de los glomérulos con lesiones segmentarias deben tener esclerosis perihiliar y/o hialinosis.
Variante celular
Al menos 1 glomérulo tiene hipercelularidad endocapilar segmentaria que obstruye la luz, con o sin células espumosas y cariorrexis.
Variante del segmento apical
Al menos 1 lesión segmentaria afecta al segmento apical (25% externo del ovillo glomerular que está junto al origen del túbulo proximal).
Variante colapsante
Al menos 1 glomérulo tiene colapso segmentario o global e hiperplasia de los podocitos suprayacentes.
Algunos investigadores creen que esta no es una forma de glomeruloesclerosis focal y segmentaria (GEFS) sino un trastorno particular denominado glomerulopatía colapsante.
Variante no especificada
Al menos 1 glomérulo tiene aumento segmentario en la matriz, lo cual oblitera el lumen capilar.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad