Abordagem
São reconhecidos quatro defeitos hereditários do metabolismo de bilirrubina. A síndrome de Gilbert e a síndrome de Crigler-Najjar (tipos I e II) estão associadas à hiperbilirrubinemia não conjugada. A síndrome de Dubin-Johnson (SDJ) e a síndrome de Rotor resultam em hiperbilirrubinemia conjugada.[28] A SDJ e a síndrome de Rotor têm uma evolução relativamente benigna.
O diagnóstico é importante para evitar investigações desnecessárias e aliviar a preocupação. Outras causas de hiperbilirrubinemia conjugada, como colestase intra-hepática recorrente benigna, podem apresentar-se com sintomas além de icterícia e mostrar evidências de dano hepático progressivo na apresentação. Isso é incomum na SDJ ou na síndrome de Rotor.
História
O diagnóstico de SDJ deve ser considerado em um paciente saudável com história de icterícia. Pode ser intermitente com exacerbações perceptíveis após infecção, doença intercorrente ou gravidez, ou durante o uso de contraceptivos orais. É postulado que a redução da função excretora hepática induzida por esteroides sexuais pode transformar hiperbilirrubinemia leve em icterícia evidente.[5][15] O estresse também pode ser um fator desencadeante.[3] Os pacientes geralmente têm alguns outros sintomas. Dor abdominal vaga e fadiga foram relatadas, embora não sejam consistentes e não estejam correlacionadas com a gravidade da patologia subjacente.[1][2][3] Diferentemente das síndromes associadas à colestase verdadeira, não há nenhum prurido.[5][6]
Tipicamente, os pacientes têm entre 10 e 30 anos quando o diagnóstico é feito.[5] Raramente, a SDJ pode se manifestar em neonatos. A SDJ é mais frequente em homens e em judeus iranianos ou marroquinos.[15][16][17] Pode haver uma história familiar positiva.
Exame
A descoloração amarela da pele, na esclera e nas membranas mucosas confirma a icterícia. Hepatomegalia pode estar presente. O exame físico geral pode revelar características de uma infecção ou doença intercorrente, mas os sinais não são específicos da SDJ. A presença de outros sinais de doença hepática, como hepatomegalia com sensibilidade ao toque, esplenomegalia, unhas brancas decorrente de hipoalbuminemia, baqueteamento digital ou hematomas frequentes, sugere uma causa alternativa de icterícia.
Achados laboratoriais
Todos os pacientes com icterícia de etiologia desconhecida devem ser submetidos a um conjunto completo de testes da função hepática (TFH) e um coagulograma. Geralmente, há elevação de bilirrubina conjugada no soro, mas outros TFHs, como aminotransferases, fosfatase alcalina e gama-glutamiltransferase, estão geralmente em níveis normais.[29] Isso ajuda a diferenciar entre SDJ e obstrução biliar extra-hepática.[30] Ocasionalmente, pode haver uma leve elevação da alanina aminotransferase (<2 vezes o limite superior do normal).[31] Normalmente, os valores de bilirrubina total estão na faixa de 2 a 5 mg/dL, com aumento da bilirrubina conjugada no plasma.[32] Os ácidos biliares séricos estão geralmente normais, contrastando com alguns distúrbios colestáticos que também se apresentam com hiperbilirrubinemia conjugada.[33] Ocasionalmente, podem ser observadas pequenas elevações nos ácidos biliares. Os tempos de coagulação não são afetados.
Investigações subsequentes
O diagnóstico é sugerido demonstrando-se um aumento na razão entre coproporfirina I e coproporfirina III urinárias. As coproporfirinas são subprodutos da biossíntese do heme. A coproporfirina I é geralmente excretada na bile, ao passo que a coproporfirina III é preferencialmente excretada na urina. Na SDJ, mais de 80% da coproporfirina excretada é do tipo I.[5][33][34]
Os níveis totais de coproporfirina podem estar elevados em pacientes com vários distúrbios hepatobiliares, mas essa alteração na razão é exclusiva da SDJ.[5][33][34] Neonatos saudáveis demonstraram ter aumentos impressionantes dos níveis de coproporfirina urinária, e mais de 80% são o isômero I durante os 2 primeiros dias de vida; contudo, no dia 10, os níveis caem para coincidir com os valores normais de adultos.[33][35] A cintilografia hepatobiliar com ácido iminodiacético marcado com 99mTc (colecintigrafia) pode ser útil quando o diagnóstico não é claro ou há suspeitas de outras doenças.[30]
Colecintigrafia
A captação do radionuclídeo 99mTc pelo fígado é excelente, mas a sua excreção do fígado para o trato biliar está prejudicada. É obtida uma colecintigrafia característica, que mostra a visualização intensa e prolongada do fígado com visualização tardia da vesícula biliar e do ducto biliar comum. O padrão é diferente daquele observado em pacientes com doença hepatocelular, obstrução extra-hepática ou síndrome de Rotor.
Biópsia hepática
A biópsia hepática percutânea é recomendada para alguns pacientes com suspeita de SDJ a fim de estabelecer o diagnóstico e descartar uma patologia mais grave do fígado. A histologia do fígado normal e a deposição no parênquima característica de um pigmento semelhante à melanina são diagnósticas. Alternativamente, um fígado escuro pode ser observado durante uma cirurgia por outra causa, o que exige uma biópsia hepática. (A coproporfirina urinária é um marcador substituto em vez de um teste definitivo. A SDJ é uma doença extremamente rara, portanto, uma investigação definitiva que confirmará o diagnóstico e descartará outras doenças será útil para tranquilizar os pacientes quanto à natureza benigna dessa doença.)
Investigações adicionais não são necessárias, mas podem ter sido realizadas como parte de uma avaliação geral de icterícia ou antes de o diagnóstico de SDJ ser feito:
Uma ultrassonografia do fígado e da árvore biliar é tipicamente normal; a colecistografia oral não visualiza a vesícula biliar, mesmo quando é realizada com doses suplementares de contraste.
Um fígado escuro observado durante a cirurgia poderá exigir a biópsia hepática para excluir uma patologia mais grave. Histologia do fígado normal e a deposição no parênquima característica de um pigmento semelhante à melanina ajudaria a estabelecer um diagnóstico de SDJ. Isso pode ser demonstrado com a coloração de Fontana-Masson ou com imuno-histoquímica. Essa pigmentação marcante de células do fígado é ausente em todas as outras doenças hepáticas, incluindo a síndrome de Rotor.[3][Figure caption and citation for the preceding image starts]: Coloração de Fontana-Masson mostrando o pigmento em um paciente com síndrome de Dubin-Johnson (SDJ)Acervo pessoal do Professor Bernard Portmann, King’s College Hospital, Londres, com permissão [Citation ends].
 [Figure caption and citation for the preceding image starts]: Imuno-histoquímica de transportador canalicular multiespecífico de ânions orgânicos em paciente, mostrando o pigmento, mas não há estrutura canalicularAcervo pessoal do Professor Bernard Portmann, King’s College Hospital, Londres, com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Imuno-histoquímica de transportador canalicular multiespecífico de ânions orgânicos em paciente, mostrando o pigmento, mas não há estrutura canalicularAcervo pessoal do Professor Bernard Portmann, King’s College Hospital, Londres, com permissão [Citation ends].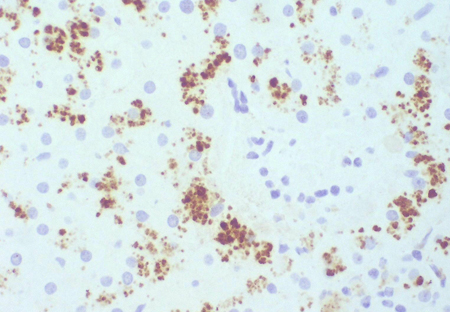 [Figure caption and citation for the preceding image starts]: Imuno-histoquímica de transportador multiespecífico de ânions orgânicos em um controle, que não mostra nenhum pigmento, mas podem ser observadas estruturas canaliculares de ramificação irregularesAcervo pessoal do Professor Bernard Portmann, King’s College Hospital, Londres, com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Imuno-histoquímica de transportador multiespecífico de ânions orgânicos em um controle, que não mostra nenhum pigmento, mas podem ser observadas estruturas canaliculares de ramificação irregularesAcervo pessoal do Professor Bernard Portmann, King’s College Hospital, Londres, com permissão [Citation ends].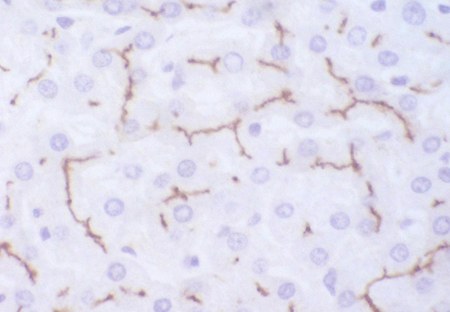
Sequenciamento do gene
Há relatos de que a análise mutacional do gene ABCC2 (que codifica a MRP2), utilizando sequenciamento de nova geração e sequenciamento de Sanger, é uma possível ferramenta de diagnóstico.[31][36][37]
Novos exames
A proteína 2 associada à resistência a múltiplos medicamentos (MRP2) também transporta leucotrienos para a bile. Quando deficiente, como na SDJ, há aumento da excreção urinária dos metabólitos de leucotrienos. Isso pode se tornar um teste diagnóstico útil, mas não está ainda em uso clínico disseminado.[38]
Alguns estudos relataram que mutações deletérias graves envolvendo os cassetes de ligação à ATP da proteína MRP2 foram mais comuns em pacientes com SDJ neonatal, enquanto variantes envolvendo outros domínios da proteína MRP2 foram mais comuns em pacientes adultos.[31][39][40]
O uso deste conteúdo está sujeito ao nosso aviso legal