Cerca de 88% a 100% dos pacientes com síndrome de Peutz-Jeghers (SPJ) desenvolvem pólipos.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Geralmente, os sintomas relacionados com pólipos surgem na infância e são observados até os 10 anos de idade em 33% dos pacientes, e até os 20 anos em 50% dos pacientes.[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
Alguns pacientes apresentam-se para uma avaliação pré-sintomática com base na história familiar da doença.
Anamnese e história familiar
Uma história de polipose intestinal, polipose extraintestinal, pigmentação mucocutânea ou neoplasias no paciente e na família deve ser observada. A polipose gástrica, do intestino delgado e colorretal ocorre em 88% a 100% dos pacientes, com a maioria aparecendo no intestino delgado (60% a 90%) e no cólon (50% a 64%).[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
O crescimento dos pólipos começa na infância, até os 10 anos de idade, e a maioria das pessoas apresenta sintomas como sangramento, dor abdominal, intussuscepção ou obstrução até os 18 anos.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
O sangramento pode ser oculto ou evidente e pode resultar em anemia ferropriva e fadiga. A pigmentação mucocutânea afeta >95% dos indivíduos e, muitas vezes, pode ter estado presente na infância, mas desaparece com o tempo.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
Exame físico
A SPJ pode ser reconhecida pelas máculas pigmentadas características (manchas planas, cinza-azuladas a marrons, com 1 a 5 milímetros) que costumam ocorrer na região perioral e na mucosa bucal (94%). Também podem ocorrer nos dedos das mãos, palmas, solas, rosto, antebraço, região perianal e, raramente, na mucosa intestinal.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
[15]Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006 Apr;4(4):408-15.
http://www.ncbi.nlm.nih.gov/pubmed/16616343?tool=bestpractice.com
As máculas nos lábios são distintas, pois cruzam a borda vermelha e, normalmente, são mais escuras e mais densamente agrupadas que as sardas.[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
As máculas pigmentadas na pele aumentam em intensidade na primeira infância e frequentemente esmaecem na vida adulta, desaparecendo completamente em alguns pacientes. As lesões bucais geralmente persistem.[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
Embora incomum, anemia pode surgir em consequência de hemorragia digestiva secundária aos pólipos; palidez, fadiga e taquicardia podem estar presentes.
Os homens devem ser examinados quanto a sinais de tumores testiculares. Testículos levemente aumentados (sem massas) ou características de puberdade precoce feminizante (por exemplo, ginecomastia bilateral) podem sugerir um tumor das células de Sertoli subjacente. A média de idade para o diagnóstico de câncer de testículo é de 9 anos, com uma faixa de 3 a 20 anos.[16]Giardiello FM, Bresinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000 Dec;119(6):1447-53.
http://www.ncbi.nlm.nih.gov/pubmed/11113065?tool=bestpractice.com
Um prolapso de pólipo perianal pode ocasionalmente estar presente em ambos os sexos.[Figure caption and citation for the preceding image starts]: Pigmentação característica presente nos lábios e na mucosa bucalDo acervo de Dra. Carol A. Burke, usado com permissão [Citation ends].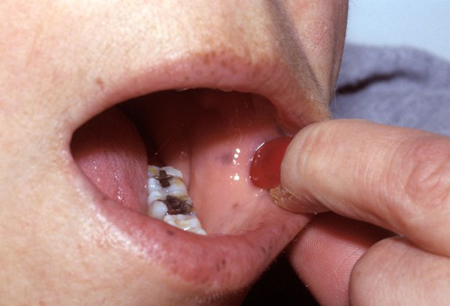
Critérios de diagnóstico
O diagnóstico de síndrome de Peutz-Jeghers (SPJ) deve ser considerado em alguém que satisfaça qualquer das condições a seguir:[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
Dois ou mais pólipos hamartomatosos do tipo Peutz-Jeghers confirmados histologicamente (pólipos do tipo PJ)
Qualquer quantidade de pólipos do tipo PJ com história familiar de SPJ em parente de primeiro grau
Pigmentação mucocutânea característica com história familiar de SPJ
Qualquer quantidade de pólipos do tipo PJ e pigmentação mucocutânea característica.
Indivíduos que atendem aos critérios clínicos devem ser submetidos a uma avaliação genética.
Teste genético
O teste genético deve ser utilizado para confirmar o diagnóstico clínico em pacientes que satisfazem os critérios de diagnóstico para SPJ.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
O sequenciamento do gene STK11 no cromossomo 19p13.3, que codifica a serino-treonina quinase LKB1, detectará 30% a 69% das mutações, e a análise de deleção/duplicação encontrará outros 30% de alterações na linha germinativa.[12]Amos CI, Keitheri-Cheteri MB, Sabripour M, et al. Genotype-phenotype correlations in Peutz-Jeghers syndrome. J Med Genet. 2004 May;41(5):327-33.
https://jmg.bmj.com/content/41/5/327.long
http://www.ncbi.nlm.nih.gov/pubmed/15121768?tool=bestpractice.com
[17]Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic STK11 deletions in Peutz-Jeghers syndrome. Hum Mutat. 2005 Dec;26(6):513-9.
http://www.ncbi.nlm.nih.gov/pubmed/16287113?tool=bestpractice.com
[18]Volikos E, Robinson J, Aittomaki K, et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006 May;43(5):e18.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2564523
http://www.ncbi.nlm.nih.gov/pubmed/16648371?tool=bestpractice.com
Um resultado positivo confirma o diagnóstico. Pequenas e grandes deleções, inserções, mutações splicing e mutações de sentido incorreto no gene STK11 foram relatadas.[2]Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998 Jan;18(1):38-43.
http://www.ncbi.nlm.nih.gov/pubmed/9425897?tool=bestpractice.com
[3]Hemminki A, Makie D, Tomlinson I, et al. A serine threonine kinase gene defect in Peutz Jeghers syndrome. Nature. 1998 Jan 8;391(6663):184-7.
http://www.ncbi.nlm.nih.gov/pubmed/9428765?tool=bestpractice.com
Um resultado negativo reduz significativamente a probabilidade de que o paciente tenha SPJ, embora mutações variantes possam ter passado despercebidas. Uma variante de significado indeterminado (VSI) também pode ser identificada.
Se um indivíduo atender aos critérios de diagnóstico clínico para SPJ, mas apresentar um resultado negativo ou indeterminado no teste genético, ele deve ser acompanhado como se tivesse SPJ ou realizar uma avaliação adicional para outra síndrome genética.[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Quando uma mutação for identificada em um probando, outros indivíduos em risco na família podem fazer o teste para esta alteração familiar específica. Deve-se reconhecer que, embora a maioria dos pacientes tenha um parente afetado, aproximadamente 25% dos pacientes apresentam mutações "de novo".[4]Syngal S, Brand RE, Church JM, et al; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb;110(2):223-62.
https://journals.lww.com/ajg/Fulltext/2015/02000/ACG_Clinical_Guideline__Genetic_Testing_and.8.aspx
http://www.ncbi.nlm.nih.gov/pubmed/25645574?tool=bestpractice.com
Pais de pacientes com tais mutações presumivelmente "de novo" devem ser cuidadosamente avaliados em busca de características de SPJ (pólipos cólicos ou pigmentação mucocutânea). Caso um dos pais seja afetado, o teste deve ser oferecido aos irmãos do probando.
Quer a mutação seja hereditária ou "de novo", todos os filhos do probando têm 50% de risco de herdá-la, e devem ser testados de acordo. Depois do aconselhamento genético adequado e de consentimento informado, o teste de membros da família em risco pode ser realizado durante a infância.
Gene reviews: Peutz-Jeghers syndrome
Opens in new window
Avaliação endoscópica
A avaliação endoscópica é indicada para três grupos de indivíduos:
Indivíduos assintomáticos com SPJ sob vigilância
Indivíduos sintomáticos ou pacientes com anormalidades laboratoriais ou radiográficas que sugiram SPJ
Parentes de primeiro grau em risco que preencham os critérios fenotípicos (isto é, pólipos cólicos ou pigmentação mucocutânea) quando não for identificada a mutação na família.
A vigilância do cólon, estômago e duodeno por endoscopia digestiva alta (EDA) e colonoscopia deve começar aos 8 anos de idade em indivíduos afetados assintomáticos (e antes naqueles que apresentam sintomas).[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
[19]Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411-44.
https://gut.bmj.com/content/69/3/411.long
http://www.ncbi.nlm.nih.gov/pubmed/31780574?tool=bestpractice.com
A vigilância do intestino delgado com videoendoscopia por cápsula (VEC) ou enterografia por ressonância magnética (ERM) também deve começar aos 8 anos de idade.[5]Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul;59(7):975-86.
http://www.ncbi.nlm.nih.gov/pubmed/20581245?tool=bestpractice.com
[13]Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):442-52.
https://journals.lww.com/jpgn/Fulltext/2019/03000/Management_of_Peutz_Jeghers_Syndrome_in_Children.31.aspx
http://www.ncbi.nlm.nih.gov/pubmed/30585892?tool=bestpractice.com
[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
[19]Monahan KJ, Bradshaw N, Dolwani S, et al. Guidelines for the management of hereditary colorectal cancer from the British Society of Gastroenterology (BSG)/Association of Coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom Cancer Genetics Group (UKCGG). Gut. 2020 Mar;69(3):411-44.
https://gut.bmj.com/content/69/3/411.long
http://www.ncbi.nlm.nih.gov/pubmed/31780574?tool=bestpractice.com
[20]van Leerdam ME, Roos VH, van Hooft JE, et al. Endoscopic management of polyposis syndromes: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy. 2019 Sep;51(9):877-95.
https://www.thieme-connect.com/products/ejournals/html/10.1055/a-0965-0605
http://www.ncbi.nlm.nih.gov/pubmed/31342472?tool=bestpractice.com
A endoscopia por cápsula é segura para o uso em indivíduos com SPJ e polipose do intestino delgado que não apresentam sintomas obstrutivos. Se houver preocupação relativa a retenção da cápsula, deve-se utilizar uma cápsula biodegradável. Em geral, os eventos adversos associados à EDA diagnóstica são raros (taxas relatadas: infecção <0.3%, sangramento <0.1%, eventos cardiopulmonares <0.1%, perfuração <0.01%).[21]ASGE Standards of Practice Committee, Coelho-Prabhu N, Forbes N, et al. Adverse events associated with EGD and EGD-related techniques. Gastrointest Endosc. 2022 Sep;96(3):389-401.e1.
https://www.giejournal.org/article/S0016-5107(22)00337-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35843754?tool=bestpractice.com
O achado endoscópico mais comum é polipose hamartomatosa difusa. A polipose gástrica, do intestino delgado e colorretal ocorre em 88% a 100% dos pacientes, com a maioria aparecendo no intestino delgado (60% a 90%) e no cólon (50% a 64%).[14]Boland CR, Idos GE, Durno C, et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: recommendations from the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology. 2022 Jun;162(7):2063-85.
https://www.gastrojournal.org/article/S0016-5085(22)00151-2/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/35487791?tool=bestpractice.com
Em casos raros, pólipos também ocorreram na pelve renal, bexiga, pulmões e narinas. Pólipos podem ser pequenos ou grandes, sésseis ou pedunculados. Eles frequentemente possuem uma aparência cerebriforme característica causada pela arborização dos músculos lisos no interior dos pólipos. Pólipos múltiplos são típicos; geralmente o número total é inferior a 20. O tamanho dos pólipos varia de alguns milímetros a vários centímetros. Patologicamente, o pólipo é de natureza hamartomatosa; o núcleo de músculo liso é coberto por lâmina própria e epitélio glandular maduro.[Figure caption and citation for the preceding image starts]: Pólipo hamartomatoso identificado no intestino delgado por endoscopia por cápsulaDo acervo de Dra. Carol A. Burke, usado com permissão [Citation ends].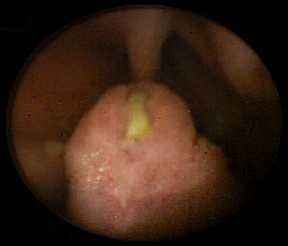 [Figure caption and citation for the preceding image starts]: Massa polipoide no intestino delgadoDo acervo do Dr. James Church, usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Massa polipoide no intestino delgadoDo acervo do Dr. James Church, usado com permissão [Citation ends].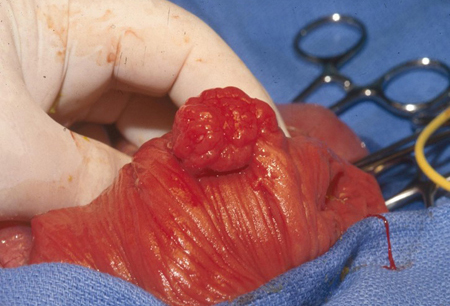 [Figure caption and citation for the preceding image starts]: Ondulação serosa característica resultante de hamartoma pedunculado no intestino delgado de paciente com síndrome de Peutz-Jeghers (SPJ)Do acervo do Dr. James Church, usado com permissão [Citation ends].
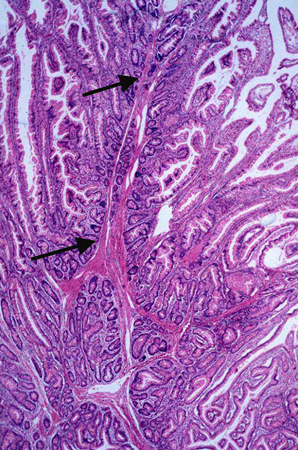
[Figure caption and citation for the preceding image starts]: Ondulação serosa característica resultante de hamartoma pedunculado no intestino delgado de paciente com síndrome de Peutz-Jeghers (SPJ)Do acervo do Dr. James Church, usado com permissão [Citation ends].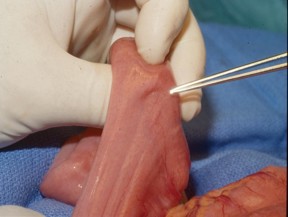 [Figure caption and citation for the preceding image starts]: Histologia de um pólipo hamartomatosoDo acervo de Dra. Carol A. Burke, usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Histologia de um pólipo hamartomatosoDo acervo de Dra. Carol A. Burke, usado com permissão [Citation ends].