Etiologia
As distonias podem ser idiopáticas, hereditárias ou adquiridas.[1]
Na distonia idiopática não há gene, lesão estrutural ou exposição responsáveis pela distonia identificados.[1][12]
Vários genes foram identificados em associação com a distonia hereditária. Estes incluem fatores autossômicos dominantes (geralmente com penetrância incompleta), autossômicos recessivos, ligados ao cromossomo X e causas genéticas mitocondriais.[12] Em decorrência de uma penetrância reduzida, a história familiar não se apresenta uniformemente nas formas genéticas de distonia.
Mutações no gene TOR1A (também conhecido como gene DYT1) que codifica TorsinA podem ser encontradas nos pacientes com distonia de início precoce (geralmente antes dos 26 anos de idade) tipicamente focal de início em um membro e, então, tornando-se generalizada.[13][14] Outras distonias isoladas podem estar associadas a mutações genéticas, incluindo uma distonia predominantemente cervical e de membros superiores com início na segunda e terceira décadas de vida, caracterizada por mutações no gene THAP1A (também conhecido como locus DYT6).[15]
Mutações no gene CGH1 estão associadas a uma distonia autossômica dominante com resposta clínica à levodopa.[16][17][Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 [Citation ends].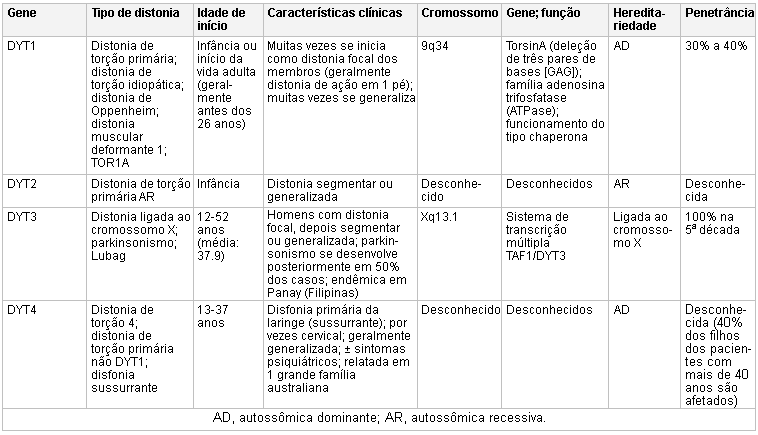 [Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 [Citation ends].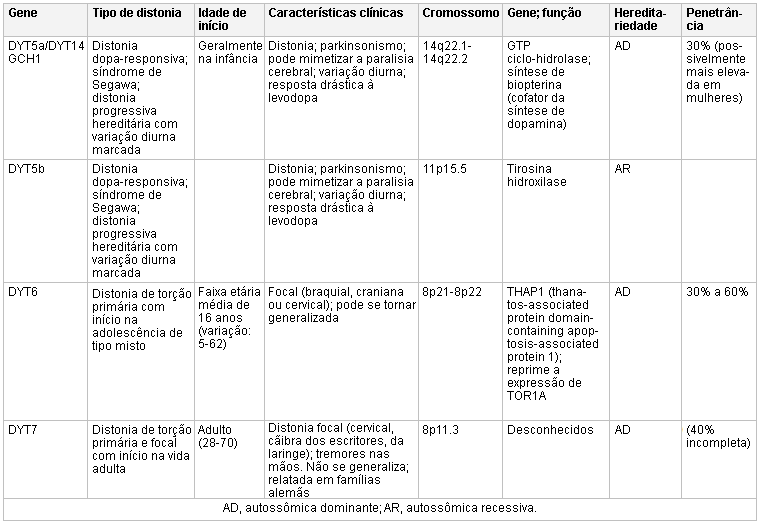 [Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 e Neuropath Applied Neurobiol. 2012 Oct;38(6):520-34 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 e Neuropath Applied Neurobiol. 2012 Oct;38(6):520-34 [Citation ends].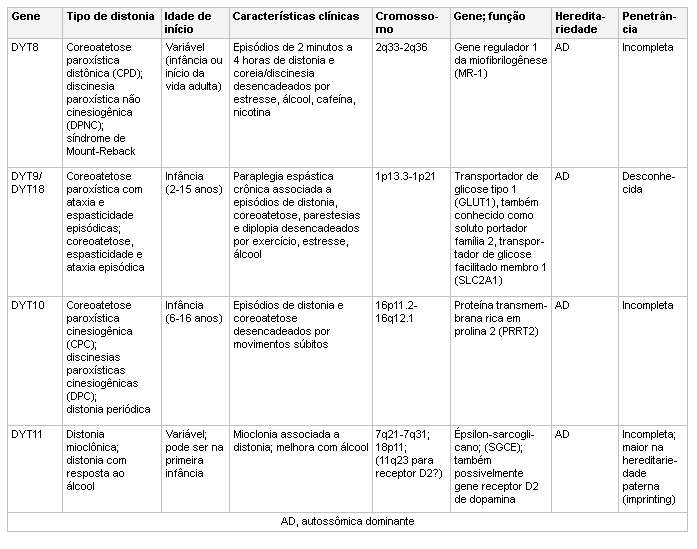 [Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 [Citation ends].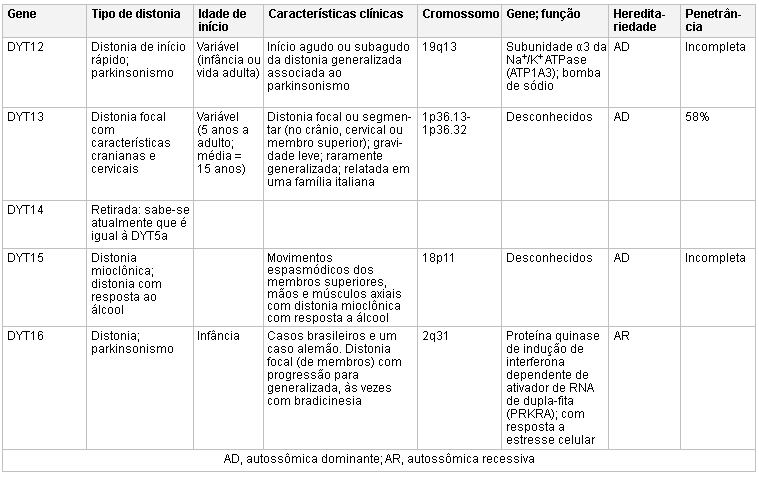 [Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 e Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genética e a distoniaAdaptado de N Engl J Med. 2006 Aug 24;355(8):818-29; usado com permissão. Informações adicionais de Mov Disord. 2011 May;26(6):1106-26 e Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].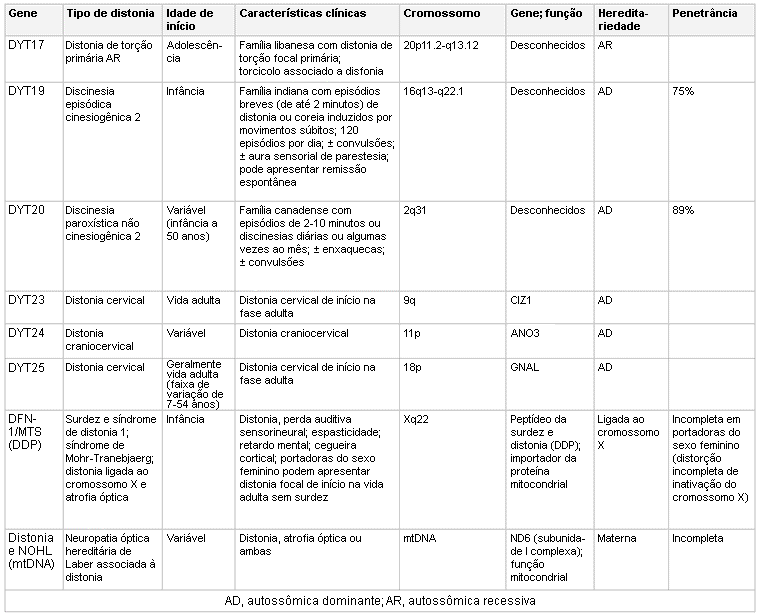
As distonias adquiridas podem ser devidas aos seguintes fatores:
Uma lesão estrutural nos gânglios da base (acidente vascular cerebral [AVC], tumor, infecção); também relatada em associação a lesões do tálamo, tronco encefálico, córtex ou cerebelo.
Uma associação a outros deficits neurológicos (por exemplo, doença de Parkinson, doença de Wilson, paralisia cerebral). Uma lesão cerebral perinatal juntamente com a ausência de desenvolvimento inicial normal é mais sugestiva de uma distonia adquirida (paralisia cerebral) do que de uma distonia idiopática.
A exposição aguda ou crônica a agentes antipsicóticos típicos e atípicos e a antieméticos bloqueadores dos receptores de dopamina, como a metoclopramida e a proclorperazina, está fortemente associada a distonias induzidas por medicamentos. Elas podem ser reações distônicas agudas ou distonias tardias.
Trauma; a distonia pós-traumática é geralmente acompanhada por outros sinais neurológicos, incluindo tremor, fraqueza e espasticidade, se a causa for um trauma cranioencefálico. Acredita-se que uma distonia fixa de início relativamente rápido, acompanhada por sinais de síndrome da dor regional complexa, compreenda uma outra forma de distonia pós-traumática, geralmente uma distonia focal de membros.[18] Essa forma de distonia geralmente não tem uma boa resposta clínica aos tratamentos usados para a distonia primária. Há diversos relatos de pacientes que desenvolveram uma distonia aparentemente focal em uma parte do corpo lesada de modo traumático há dias ou semanas, embora a relação etiológica exata entre o trauma e a distonia ainda seja controversa.[19][20][21]
Atividade; há fortes evidências anedóticas sugerindo que pessoas que frequentemente usam habilidades motoras finas (por exemplo, músicos) apresentam aumento do risco de desenvolvimento de distonia focal.
A distonia também pode ser uma característica de um distúrbio neurodegenerativo disseminado complexo, também conhecido como distonia heredodegenerativa.[3][22][23][24][25] A distonia funcional e a distonia paroxística também são subtipos de distonia reconhecidos.[2][26]
Fisiopatologia
A fisiopatologia é complexa, com fatores distintos contribuindo para o desenvolvimento da distonia em diferentes condições. A fisiopatologia exata das distonias idiopáticas ainda não é conhecida. É provável que a dopamina afete alguns tipos de distonia, conforme sugerido pela deficiência de dopamina e pela drástica resposta a agentes dopaminérgicos na distonia dopa-responsiva. A associação frequente da distonia ao parkinsonismo e a indução de distonia por medicamentos antipsicóticos bloqueadores de dopamina são outras evidências de haver uma participação da dopamina na distonia.[3][23]
Está cada vez mais claro que as formas focais e generalizadas de distonia são distúrbios da rede cerebral que podem resultar na perda da inibição normal de áreas motoras, integração motora e sensorial anormal e plasticidade mal-adaptativa.[27] Múltiplos estudos de neuroimagem demonstraram padrões anormais de atividade metabólica e alterações microestruturais nos núcleos da base, tálamo, cerebelo e córtices sensoriomotor e pré-motor.[27][28] A tomografia por emissão de pósitrons com fluordeoxiglicose e os estudos de ressonância nuclear magnética funcional geralmente mostram aumento no metabolismo da glicose em repouso no córtex pré-motor e no núcleo lentiforme, bem como atividade possivelmente anormal nos córtices pré-motor e motor primário. Deve-se notar que esses exames de imagem do cérebro não são usados para confirmar um diagnóstico clínico de distonia. Estudos eletrofisiológicos indicam comprometimento da atividade inibitória do sistema nervoso central. O comprometimento da inibição associada pode contribuir para a disseminação da atividade neural em regiões adjacentes aos circuitos neurais ativados, sendo potencialmente o causador do movimento que se espalha aos músculos adjacentes (fenômeno de overflow). As representações motoras e sensoriais das partes do corpo afetadas na distonia focal são aumentadas no córtex cerebral. No entanto, ainda não está claro se essas alterações são primárias ou secundárias.[23][29]
Classificação
Classificação da distonia: características clínicas e etiologia[1]
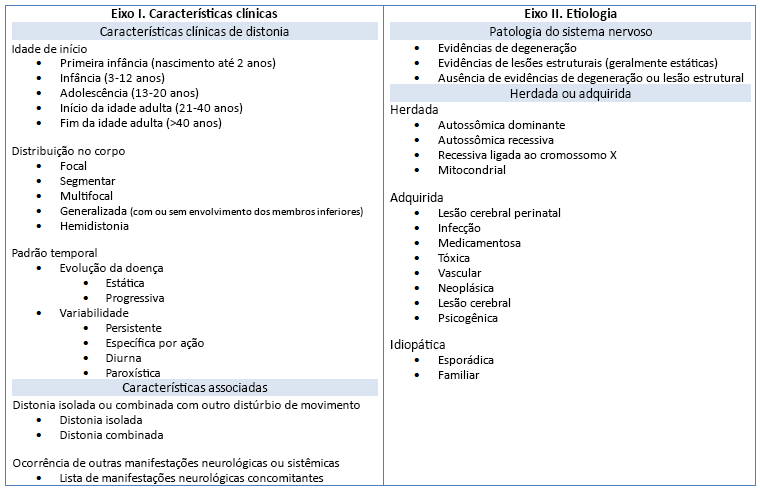
Novas informações sobre etiologia, e problemas com a terminologia prévia, levaram ao desenvolvimento e publicação de uma classificação atualizada de distonias por um consenso do comitê de especialistas. As distonias agora são classificadas de acordo com dois eixos principais: manifestações clínicas e causas biológicas.
Eixo I. Características clínicas
A classificação dicotômica das síndromes de distonia (com início na infância ou na idade adulta) foi substituída por um esquema que classifica as distonias em cinco faixas etárias (primeira infância, infância, adolescência, início da idade adulta e idade adulta posterior).
As distribuições corporais afetadas incluem a focal, a segmentar, a multifocal, a generalizada e a hemidistonia.
Padrões temporais exibidos nas distonias. O ciclo da doença pode ser estático ou progressivo, enquanto a variabilidade da doença inclui padrões persistentes, de ações específicas, diurnos e paroxísticos.
As características associadas são importantes. A distonia pode ocorrer isoladamente, ou pode haver distúrbios do movimento adicionais. A distonia isolada inclui distonias previamente chamadas de distonia "primária", em que, com exceção do tremor, a distonia é a única anomalia motora. A distonia combinada inclui distonias associadas a outros distúrbios do movimento, como parkinsonismo ou mioclonia. Além disso, pode haver outras manifestações neurológicas ou sistêmicas associadas à distonia.
Na classificação do consenso de 2013, "isolada" refere-se à fenomenologia e não à etiologia. Nas formas "combinadas", a distonia não é necessariamente o distúrbio do movimento predominante.
Eixo II. Etiologia
Inclui distúrbios hereditários e adquiridos nos quais há patologias neurológicas degenerativas, lesões cerebrais estruturais adquiridas, ou distúrbios sem anormalidades cerebrais identificáveis.
Distúrbios hereditários incluem doenças autossômica dominante, autossômica recessiva, ligada ao cromossomo X e mitocondriais.
As distonias hereditárias incluem um grupo misto de síndromes distônicas isoladas e combinadas, algumas das quais são muito raras.
A distonia pode ser devida a um distúrbio adquirido, como uma causa traumática, infecciosa, induzida por medicamentos, tóxica, vascular ou neoplásica, lesão cerebral perinatal ou um distúrbio do movimento funcional (psicogênico).[2]
Distonias com início na idade adulta idiopáticas focais ou segmentais com distribuição tanto familiar quanto esporádica também estão incluídas no eixo II.[Figure caption and citation for the preceding image starts]: Classificação de consenso de distoniaReimpressão com permissão de Albanese et al. Mov Disord. 2013 Jun 15;28(7):863-73 [Citation ends].
O uso deste conteúdo está sujeito ao nosso aviso legal