Etiology
Dystonias may be idiopathic, inherited, or acquired.[1]
In idiopathic dystonia there is no identified gene, structural lesion or exposure responsible for the dystonia.[1][12]
Several genes have been identified in association with inherited dystonia. These include autosomal-dominant (often with incomplete penetrance), autosomal-recessive, X-linked, and mitochondrial genetic causes.[12] A family history is not uniformly present in genetic forms of dystonia due to reduced penetrance.
Mutations in the TOR1A gene (also known as the DYT1 gene) that encodes TorsinA can be found in patients with early-onset dystonia (usually beginning before 26 years of age) that typically begins focally in one limb and subsequently generalizes.[13][14] Other isolated dystonias can be associated with genetic mutations, including a predominantly neck and upper limb dystonia with onset in the second and third decades of life that is characterized by mutations in the THAP1 gene (also known as the DYT6 locus).[15]
Mutations in the CGH1 gene are associated with an autosomal dominant, levodopa responsive dystonia.[16][17][Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 [Citation ends].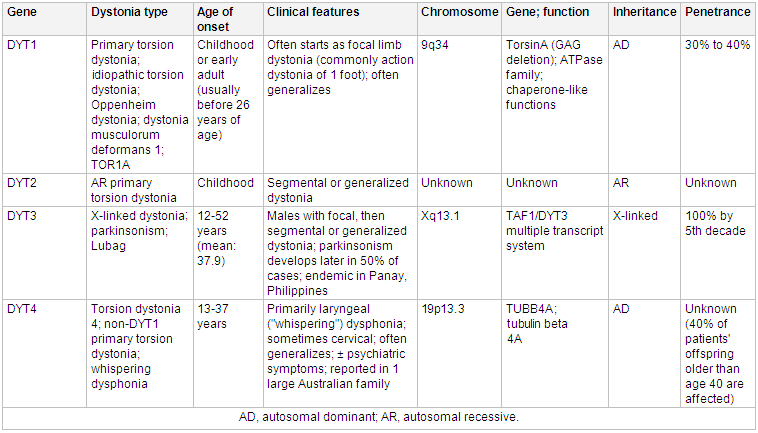 [Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 [Citation ends].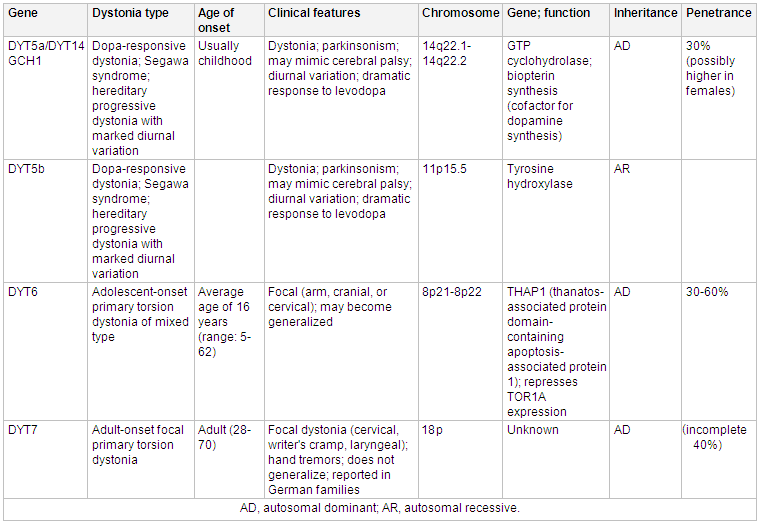 [Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information from Mov Disord. 2011 May;26(6):1106-26 and Neuropath Applied Neurobiol. 2012 Oct;38(6):520-34 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information from Mov Disord. 2011 May;26(6):1106-26 and Neuropath Applied Neurobiol. 2012 Oct;38(6):520-34 [Citation ends].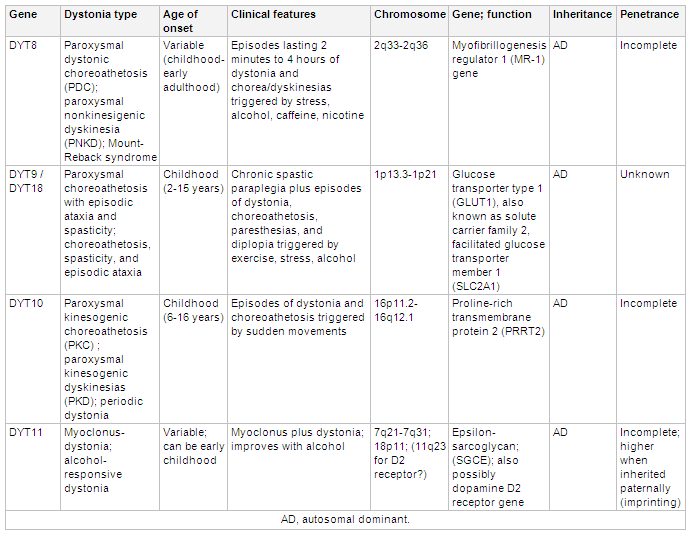 [Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 [Citation ends]. [Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 and Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].
[Figure caption and citation for the preceding image starts]: Genetics and dystoniaAdapted from N Engl J Med. 2006 Aug 24;355(8):818-29; used with permission. Additional information Mov Disord. 2011 May;26(6):1106-26 and Mov Disord. 2013 Jun 15;28(7):899-905 [Citation ends].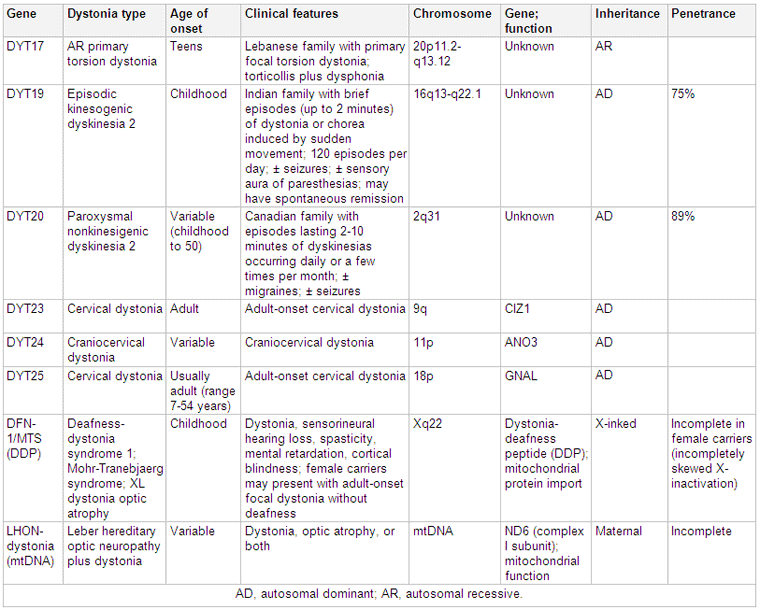
Acquired dystonias may be due to the following factors:
A structural lesion involving the basal ganglia (stroke, tumor, infection); also reported with lesions of the thalamus, brain stem, cortex, or cerebellum.
An association with other neurologic deficits (e.g., Parkinson disease, Wilson disease, cerebral palsy). Perinatal cerebral injury along with absence of normal early development is more suggestive of an acquired dystonia (cerebral palsy) rather than a idiopathic dystonia.
Acute or chronic exposure to both typical and atypical antipsychotic agents and dopamine-receptor blocker antiemetics such as metoclopramide and prochlorperazine are strongly associated with medication-induced dystonias. These can either be acute dystonic reactions or tardive dystonias.
Trauma; posttraumatic dystonia is usually accompanied by other neurologic signs including tremor, weakness, and spasticity if head trauma is the cause. A relatively rapid-onset fixed dystonia accompanied by signs of complex regional pain syndrome is thought to comprise another form of posttraumatic dystonia, usually a focal limb dystonia.[18] This form of dystonia is usually poorly responsive to the treatments used for primary dystonia. There are many reports of patients who have developed what appears to be focal dystonia of a body part that had been traumatically injured days or weeks earlier, although the precise etiologic relationship between trauma and dystonia remains controversial.[19][20][21]
Activity; there is strong anecdotal evidence suggesting that people who frequently use fine motor skills (e.g., musicians) are at increased risk of developing focal dystonia.
Dystonia also can be a feature of a more complex widespread neurodegenerative disorder, sometimes referred to as "heredodegenerative" dystonia.[3][22][23][24][25] Functional dystonia and paroxysmal dystonia are also recognized subtypes of dystonia.[2][26]
Pathophysiology
The pathophysiology is complex, with distinct factors contributing to the development of dystonia in different conditions. The precise pathophysiology of idiopathic dystonias remains unknown. Dopamine is likely to be involved in some types of dystonia, as suggested by the dopamine deficiency and dramatic responsiveness to dopaminergic agents in dopa-responsive dystonia. The frequent association of dystonia with parkinsonism and the induction of dystonia by dopamine-blocking antipsychotic medications provide further evidence of a role for dopamine in dystonia.[3][23]
It is increasingly clear that both focal and generalized forms of dystonia are brain network disorders that may result in loss of normal surround inhibition of motor areas, abnormal sensorimotor integration, and maladaptive plasticity.[27] Multiple neuroimaging studies have demonstrated abnormal patterns of metabolic activity and microstructural changes in the basal ganglia, thalamus, cerebellum, and sensorimotor and premotor cortices.[27][28] Fluoro-deoxyglucose positron-emission tomography and functional magnetic resonance imaging studies have generally shown increased resting glucose metabolism in premotor cortex and lentiform nucleus as well as possibly abnormal activity in premotor and primary motor cortex. It should be noted that these brain scans are not used to confirm a clinical diagnosis of dystonia. Electrophysiologic studies indicate impaired central nervous system inhibitory activity. Impaired surround inhibition may contribute to spread of neural activity to regions adjacent to activated neural circuits, potentially accounting for inappropriate overflow of movement into adjacent muscles. Sensorimotor representations of affected body parts in focal dystonia are enlarged in the cerebral cortex. However, it remains unclear if these changes are primary or secondary.[23][29]
Classification
Dystonia classification: clinical characteristics and etiology[1]
New information concerning etiology, and problems with older terminology, has led to the development and publication of an updated classification of dystonias by a consensus committee of experts. The dystonias are now classified according to two main axes: clinical manifestations and biological causes.
Axis I. Clinical characteristics
Dichotomous classification of dystonia syndromes (childhood- or adult-onset) has been replaced by a scheme that classifies dystonias into five age ranges (infancy, childhood, adolescence, early adulthood, and late adulthood).
Affected body distribution includes focal, segmental, multifocal, generalized, and hemidystonia.
Temporal patterns exhibited in the dystonias. The disease course may be static or progressive, while disease variability includes persistent, action-specific, diurnal, and paroxysmal patterns.
Associated features are important. Dystonia may occur in isolation, or there may be additional movement disorders. Isolated dystonia includes dystonias previously called "primary" dystonia where, with the exception of tremor, dystonia is the only motor abnormality. Combined dystonia includes dystonias associated with other movement disorders such as parkinsonism or myoclonus. In addition, there may be other neurologic or systemic manifestations that are associated with the dystonia.
In the 2013 consensus classification, "isolated" refers to phenomenology and not etiology. In "combined" forms, dystonia is not necessarily the predominant movement disorder.
Axis II. Etiology
Includes inherited and acquired disorders in which there are degenerative brain pathologies, acquired structural brain lesions, or disorders without identifiable brain abnormalities.
Inherited disorders include autosomal-dominant, autosomal-recessive, X-linked, and mitochondrial diseases.
The inherited dystonias include a mixed group of both isolated and combined dystonic syndromes, some of which are very rare.
Dystonia may be due to an acquired disorder, such as a traumatic, infectious, drug-induced, toxic, vascular, or neoplastic cause, perinatal brain injury, or a functional (psychogenic) movement disorder.[2]
Idiopathic adult-onset focal or segmental dystonias with either sporadic or familial distribution are also included in Axis II.[Figure caption and citation for the preceding image starts]: Consensus classification of dystoniaReprinted with permission from Albanese et al. Mov Disord. 2013 Jun 15;28(7):863-73 [Citation ends].

Use of this content is subject to our disclaimer