The underlying pathophysiology of Brugada syndrome (BrS) remains unclear.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
It is likely that there are a number of contributing factors that lead to the typical Brugada pattern on ECG, rather than a single mechanism, because of the range of clinical manifestations and phenocopies.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
Around 20% to 30% of people with BrS have identifiable inherited mutations of the SCN5A gene, which causes dysfunctional cardiac voltage gated NaV1.5 sodium channels.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[7]Zeppenfeld K, Tfelt-Hansen J, de Riva M, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022 Oct 21;43(40):3997-4126.
https://academic.oup.com/eurheartj/article/43/40/3997/6675633?login=false
http://www.ncbi.nlm.nih.gov/pubmed/36017572?tool=bestpractice.com
[16]Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010 Jan;7(1):33-46.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2822446
http://www.ncbi.nlm.nih.gov/pubmed/20129283?tool=bestpractice.com
[17]Hu D, Barajas-Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64(1):66-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4116276
http://www.ncbi.nlm.nih.gov/pubmed/24998131?tool=bestpractice.com
[18]Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm. 2018 Oct;15(10):e190-e252.
https://www.heartrhythmjournal.com/article/S1547-5271(17)31249-3/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/29097320?tool=bestpractice.com
These defective sodium channels shorten the duration of the cardiac action potential, leading to a reduction of the peak influx of sodium ions and a slowing in the upstroke (phase 0) of the cardiac action potential.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
Over 300 mutations of the SCN5A gene have been discovered so far.[22]Attard A, Stanniland C, Attard S,et al. Brugada syndrome: should we be screening patients before prescribing psychotropic medication? Ther Adv Psychopharmacol. 2022 Jan 28;12:20451253211067017.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8801628
http://www.ncbi.nlm.nih.gov/pubmed/35111298?tool=bestpractice.com
This range of genetic mutations may cause differences in electrophysiological abnormalities, which could explain the different clinical manifestations of BrS.[23]Deschênes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46(1):55-65.
https://academic.oup.com/cardiovascres/article/46/1/55/333556?login=true
http://www.ncbi.nlm.nih.gov/pubmed/10727653?tool=bestpractice.com
[24]Clancy CE, Kass RS. Defective cardiac ion channels: from mutations to clinical syndromes. J Clin Invest. 2002;110(8):1075-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC150807
http://www.ncbi.nlm.nih.gov/pubmed/12393842?tool=bestpractice.com
Experimental studies have shown that the function of NaV1.5 sodium channels is also affected by changes in temperature, with additional shortening of the cardiac action potential at higher temperatures.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Cardiac action potential in Brugada syndrome. Blue line indicates normal ventricular action potential; red line indicates delayed upstroke of action potential in Brugada syndrome. Action potential phases: 0, rapid depolarisation; 1, rapid/early repolarization; 2, plateau; 3, terminal repolarisation; 4, resting potentialKrahn AD et al. JACC Clin Electrophysiol 2022 Mar;8(3):386-405; used with permission [Citation ends].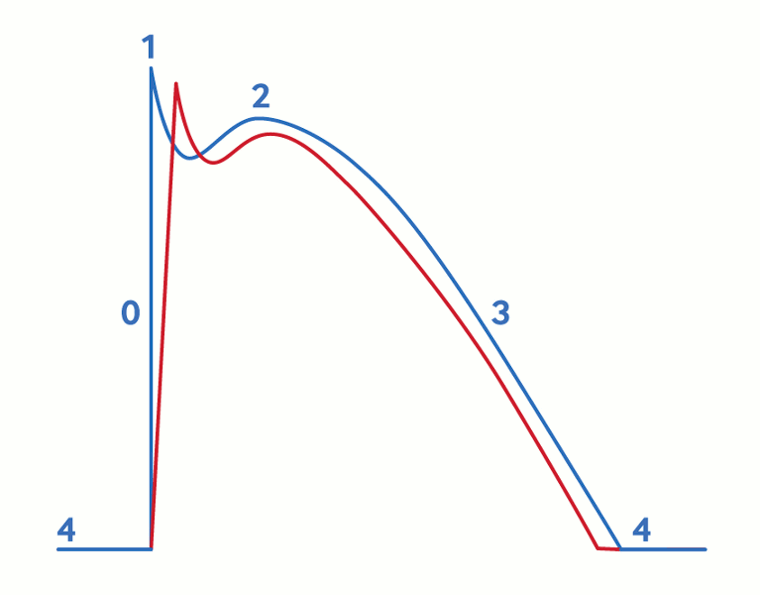
Other genes that have been implicated in BrS (although their significance is disputed; only SCN5A gene variants are considered definitely disease-causing) include SCN10A (which encodes for the a-subunit of the NaV1.8 sodium channel), those that encode for NaV1.5 b-subunits, those involved in NaV1.5 trafficking or expression, and potassium and calcium channel genes.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[17]Hu D, Barajas-Martínez H, Pfeiffer R, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. 2014;64(1):66-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4116276
http://www.ncbi.nlm.nih.gov/pubmed/24998131?tool=bestpractice.com
[19]Hosseini SM, Kim R, Udupa S, etal. Reappraisal of reported genes for sudden arrhythmic death: Evidence-based evaluation of gene validity for Brugada syndrome. Circulation. 2018 Sep 18;138(12):1195-205.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6147087
http://www.ncbi.nlm.nih.gov/pubmed/29959160?tool=bestpractice.com
[25]Behr ER, Savio-Galimberti E, Barc J, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res. 2015;106(3):520-9.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4447806
http://www.ncbi.nlm.nih.gov/pubmed/25691538?tool=bestpractice.com
[26]Fukuyama M, Ohno S, Makiyama T, Horie M. Novel SCN10A variants associated with Brugada syndrome. Europace. 2016;18(6):905-11.
https://academic.oup.com/europace/article/18/6/905/2467006?login=false
http://www.ncbi.nlm.nih.gov/pubmed/25842276?tool=bestpractice.com
[27]Riuró H, Beltran-Alvarez P, Tarradas A, et al. A missense mutation in the sodium channel β2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat. 2013;34(7):961-6.
http://www.ncbi.nlm.nih.gov/pubmed/23559163?tool=bestpractice.com
[28]Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129(10):1092-103.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3954430
http://www.ncbi.nlm.nih.gov/pubmed/24352520?tool=bestpractice.com
[29]Belbachir N, Portero V, Al Sayed ZR, et al. RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome. Eur Heart J. 2019 Oct 1;40(37):3081-94.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6769825
http://www.ncbi.nlm.nih.gov/pubmed/31114854?tool=bestpractice.com
[30]Giudicessi JR, Ye D, Tester DJ, et al. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm. 2011;8(7):1024-32.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3150551
http://www.ncbi.nlm.nih.gov/pubmed/21349352?tool=bestpractice.com
[31]Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7(12):1872-82.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2999985
http://www.ncbi.nlm.nih.gov/pubmed/20817017?tool=bestpractice.com
There is debate about whether BrS is due to a primary repolarisation disorder, a primary depolarisation disorder, or both.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[32]Behr ER, Ben-Haim Y, Ackerman MJ, et al. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: a final common pathway? Eur Heart J. 2021 Mar 14;42(11):1073-81.
https://academic.oup.com/eurheartj/article/42/11/1073/6076711?login=false
http://www.ncbi.nlm.nih.gov/pubmed/33421051?tool=bestpractice.com
[33]Wilde AA, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49(4):543-53.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2932806
http://www.ncbi.nlm.nih.gov/pubmed/20659475?tool=bestpractice.com
The argument for a primary repolarisation disorder is supported by the fact that patients with BrS have a conduction delay in the right ventricular outflow tract, which is associated with late action potentials.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
This conduction delay results in heterogeneity of depolarisation around the right ventricular outflow tract, which is thought to predispose the patient to arrhythmias.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
The argument for a primary depolarisation disorder is supported by evidence that has shown a dispersion of transmural (epicardial-endocardial gradient) action potentials in canine models of pharmacologically induced BrS.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[33]Wilde AA, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49(4):543-53.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2932806
http://www.ncbi.nlm.nih.gov/pubmed/20659475?tool=bestpractice.com
[34]Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-6.
https://www.ahajournals.org/doi/10.1161/01.cir.100.15.1660?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/10517739?tool=bestpractice.com
Heterogeneity of repolarisation between the epicardium and endocardium is thought to cause arrhythmias by phase 2 re-entry.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[34]Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660-6.
https://www.ahajournals.org/doi/10.1161/01.cir.100.15.1660?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/10517739?tool=bestpractice.com
In addition, both repolarisation and depolarisation abnormalities have been demonstrated in people with BrS, although it is suggested that these repolarisation changes occur secondary to a primary depolarisation disorder.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
Some studies have shown that people with BrS have functional changes in the epicardial aspect of the right ventricle.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[35]Iacoviello M, Forleo C, Puzzovivo A, et al. Altered two-dimensional strain measures of the right ventricle in patients with Brugada syndrome and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur J Echocardiogr. 2011;12(10):773-81.
http://www.ncbi.nlm.nih.gov/pubmed/21865227?tool=bestpractice.com
[36]Bastiaenen R, Cox AT, Castelletti S, et al. Late gadolinium enhancement in Brugada syndrome: A marker for subtle underlying cardiomyopathy? Heart Rhythm. 2017 Apr;14(4):583-9.
http://www.ncbi.nlm.nih.gov/pubmed/27919765?tool=bestpractice.com
There have also been histological changes identified in the anterior right ventricular outflow tract that are associated with areas of low voltage and the presence of abnormal fractionated electrograms on electrophysiological studies.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
These changes include increased collagen and fibrosis, presence of inflammatory infiltrates, and a reduction in connexin-43 (a gap junction protein that provides electrical coupling between myocytes).[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[37]Pieroni M, Notarstefano P, Oliva A, et al. Electroanatomic and pathologic right ventricular outflow tract abnormalities in patients with Brugada syndrome. J Am Coll Cardiol. 2018 Dec 4;72(22):2747-57.
https://www.sciencedirect.com/science/article/pii/S0735109718386637?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/30497561?tool=bestpractice.com
[38]Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123(12):1270-9.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.110.972612?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/21403098?tool=bestpractice.com
Ablation that is targeted to these areas can result in correction of the typical Brugada pattern ECG changes and prevention of ventricular arrhythmias.[1]Krahn AD, Behr ER, Hamilton R, et al. Brugada syndrome. JACC Clin Electrophysiol. 2022 Mar;8(3):386-405.
https://www.sciencedirect.com/science/article/pii/S2405500X2101080X?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/35331438?tool=bestpractice.com
[22]Attard A, Stanniland C, Attard S,et al. Brugada syndrome: should we be screening patients before prescribing psychotropic medication? Ther Adv Psychopharmacol. 2022 Jan 28;12:20451253211067017.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8801628
http://www.ncbi.nlm.nih.gov/pubmed/35111298?tool=bestpractice.com
[37]Pieroni M, Notarstefano P, Oliva A, et al. Electroanatomic and pathologic right ventricular outflow tract abnormalities in patients with Brugada syndrome. J Am Coll Cardiol. 2018 Dec 4;72(22):2747-57.
https://www.sciencedirect.com/science/article/pii/S0735109718386637?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/30497561?tool=bestpractice.com
[38]Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123(12):1270-9.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.110.972612?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/21403098?tool=bestpractice.com
