Etiologia
A etiologia exata é desconhecida, mas parece estar relacionada a uma deficiência na capacidade de armazenamento intracelular do fígado para ânions orgânicos, incluindo diglicuronídeo de bilirrubina.[8][9]
Uma análise de 8 famílias com síndrome de Rotor (RS) descobriu que a síndrome estava associada a mutações que presumivelmente causam deficiências completas e simultâneas do ânion orgânico que transporta os polipeptídeos OATP1B1 e OATP1B3. Essas importantes proteínas limitadoras da detoxificação fazem mediação da captação e clearance de inúmeros medicamentos e medicamentos conjugados na membrana sinusoidal dos hepatócitos. O estudo sugeriu a possibilidade de toxicidade com risco de vida com certos medicamentos na RS.[10]
São reconhecidos quatro defeitos hereditários do metabolismo da bilirrubina: a síndrome de Gilbert e a síndrome de Crigler-Najjar (tipos I e II) estão associadas a hiperbilirrubinemia não conjugada, e a síndrome de Dubin-Johnson (SDJ) e a RS estão associadas a hiperbilirrubinemia conjugada. A bilirrubina é o subproduto do catabolismo de heme. [Figure caption and citation for the preceding image starts]: Representação esquemática do metabolismo da bilirrubina (B) dentro do hepatócitoDo acervo de Dr. Coelho [Citation ends].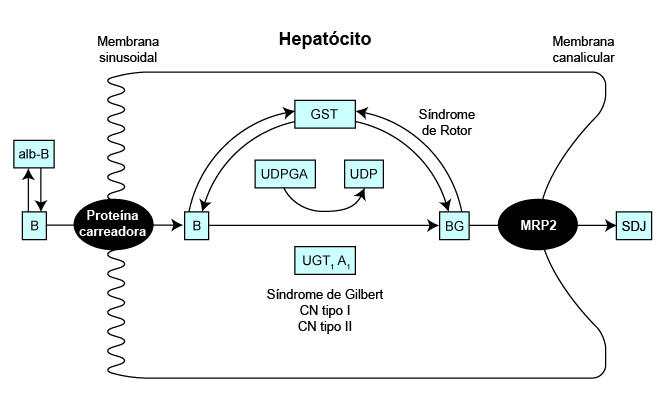 A disposição normal de bilirrubina (B) envolve seu transporte para o fígado por meio de um portador de albumina (alb-B). No hepatócito, ela é conjugada com a molécula de açúcar, o ácido glicurônico (G), para formar glicuronato de bilirrubina (BG), um composto hidrossolúvel que pode ser facilmente excretado nas fezes. O doador é uridina difosfato ácido glicurônico (UDPGA), e a reação é catalisada pela enzima uridina difosfato glicuronil transferase (isoforma específica de UGT1A1). Uma diminuição na atividade dessa enzima está associada à síndrome de Gilbert e à síndrome de Crigler-Najjar (tipos I e II). A glutationa-S-transferase (GST) age como uma proteína intracelular carreadora de certas moléculas orgânicas.[11] Sugeriu-se que os pacientes com RS podem ter uma deficiência de GST hepática.[12] Uma deficiência resultaria na captação comprometida da bilirrubina dentro do citosol. Além disso, os conjugados de bilirrubina se ligam à GST enquanto aguardam a excreção dos hepatócitos para o lúmen canalicular, portanto as deficiências no armazenamento intracelular ocasionarão o vazamento de conjugados de bilirrubina de volta para a circulação.[13] A proteína canalicular de resistência a múltiplos medicamentos 2 (MRP2) parece ser importante para a secreção canalicular da bilirrubina. Um defeito na MRP2 resulta em SDJ.
A disposição normal de bilirrubina (B) envolve seu transporte para o fígado por meio de um portador de albumina (alb-B). No hepatócito, ela é conjugada com a molécula de açúcar, o ácido glicurônico (G), para formar glicuronato de bilirrubina (BG), um composto hidrossolúvel que pode ser facilmente excretado nas fezes. O doador é uridina difosfato ácido glicurônico (UDPGA), e a reação é catalisada pela enzima uridina difosfato glicuronil transferase (isoforma específica de UGT1A1). Uma diminuição na atividade dessa enzima está associada à síndrome de Gilbert e à síndrome de Crigler-Najjar (tipos I e II). A glutationa-S-transferase (GST) age como uma proteína intracelular carreadora de certas moléculas orgânicas.[11] Sugeriu-se que os pacientes com RS podem ter uma deficiência de GST hepática.[12] Uma deficiência resultaria na captação comprometida da bilirrubina dentro do citosol. Além disso, os conjugados de bilirrubina se ligam à GST enquanto aguardam a excreção dos hepatócitos para o lúmen canalicular, portanto as deficiências no armazenamento intracelular ocasionarão o vazamento de conjugados de bilirrubina de volta para a circulação.[13] A proteína canalicular de resistência a múltiplos medicamentos 2 (MRP2) parece ser importante para a secreção canalicular da bilirrubina. Um defeito na MRP2 resulta em SDJ.
A RS é hereditária como um traço autossômico recessivo. Em um estudo, a RS era coexistente com outros distúrbios hereditários (deficiência de glicose-6-fosfato desidrogenase [G6PD] e talassemia beta heterozigótica), sugerindo uma possível interação entre genes herdados juntos.[7]
Fisiopatologia
Estudos de excreção de contraste que usam sulfobromoftaleína (BSP) mostraram que, na RS, a capacidade de transporte do contraste para a bile é reduzida em <50%. Além disso, a capacidade de armazenamento nos hepatócitos é reduzida mais de 5 vezes em comparação com valores normais.[8]
A deficiência do armazenamento e da excreção de bilirrubina e de outros ânions orgânicos leva à secreção defeituosa da bilirrubina conjugada na bile, o que resulta em sua reabsorção no sangue e excreção na urina. A circulação e o acúmulo em excesso da bilirrubina (hiperbilirrubinemia) resultam em icterícia. Ao contrário da síndrome de Dubin-Johnson, não há pigmentação hepática.
O uso deste conteúdo está sujeito ao nosso aviso legal