Abordaje
Se han reconocido cuatro defectos hereditarios del metabolismo de la bilirrubina. El síndrome de Gilbert y el síndrome de Crigler-Najjar (tipo I y II) están asociados con la hiperbilirrubinemia no conjugada. El síndrome de Dubin-Johnson (SDJ) y el síndrome de Rotor provocan hiperbilirrubinemia conjugada.[28] Tanto el síndrome de DJS como el síndrome de Rotor presentan una evolución relativamente benigna.
El diagnóstico es importante para evitar investigaciones innecesarias y aliviar la preocupación. Otras causas de hiperbilirrubinemia conjugada, como la colestasis intrahepática recurrente benigna, pueden presentarse con síntomas además de ictericia y mostrar evidencia de daño hepático progresivo en el momento de la presentación. Esto es inusual en DJS o en el síndrome de Rotor.
Antecedentes
El diagnóstico de (SDJ) debe considerarse en un paciente sano con antecedentes de ictericia. Puede ser intermitente, con exacerbaciones notorias seguidas de infección, enfermedad intercurrente o embarazo, o durante la utilización de anticonceptivos orales. Se postula que la reducción de la función excretora hepática inducida por esteroides sexuales puede transformar una hiperbilirrubinemia leve en una ictericia manifiesta.[5][15] El estrés también es un factor desencadenante posible.[3] Los pacientes suelen presentar pocos otros síntomas. Se han informado síntomas de dolor abdominal difuso y fatiga, si bien no son persistentes y no se correlacionan con la gravedad de la patología subyacente.[1][2][3] Al contrario de los casos de síndromes asociados con una colestasis verdadera, no se presenta prurito.[5][6]
Los pacientes suelen tener entre 10 y 30 años de edad al momento de diagnosticarse la enfermedad.[5] En raras ocasiones, el SDJ puede presentarse en neonatos. El SDJ es más frecuente en hombres y entre judíos de Irán y judíos de Marruecos.[15][16][17] Podrían existir antecedentes familiares positivos.
Exploración física
La coloración amarilla de la piel, la esclerótica y membranas mucosas confirman la ictericia. Puede presentarse hepatomegalia. Un examen general puede revelar características de una enfermedad o infección intercurrente, pero los signos no son específicos del SDJ. La presencia de otros signos de hepatopatía, como hepatomegalia sensible a la palpación, esplenomegalia, uñas blancas debido a hipoalbuminemia, achatamiento o tendencia a la formación de hematomas sugiere una causa alternativa para la ictericia.
Hallazgos de laboratorio
Todos los pacientes con ictericia de etiología desconocida deben someterse a una serie completa de pruebas de función hepática (PFH) y a un perfil de coagulación. Generalmente se presenta un aumento de la bilirrubina conjugada sérica, pero otras PFH como aminotransferasas, fosfatasa alcalina y gamma glutamil transferasa suelen resultar normales.[29] Esto ayuda a diferenciar entre el SDJ y una obstrucción biliar extrahepática.[30] Ocasionalmente, puede haber una leve elevación de la alanina aminotransferasa (<2 veces el límite superior de la normalidad).[31] Normalmente, los valores de bilirrubina total están en el rango de 2 a 5 mg/dL, con un aumento de la bilirrubina conjugada en plasma.[32] Los ácidos biliares séricos generalmente son normales, en contraste con algunos trastornos colestásicos que también están presentes en el caso de la hiperbilirrubinemia conjugada.[33] Ocasionalmente, se pueden observar leves aumentos en los ácidos biliares. Los tiempos de coagulación no se ven afectados.
Investigaciones posteriores
El diagnóstico se sugiere mediante la demostración de un aumento en la proporción entre la coproporfirina I y la coproporfirina III en orina. Las coproporfirinas son subproductos de la biosíntesis del hemo. La coproporfirina I generalmente es excretada en la bilis, mientras que la coproporfirina III es excretada preferentemente en la orina. En el SDJ, más del 80% de la coproporfirina excretada es de tipo I.[5][33][34]
Los niveles totales de coproporfirina pueden aumentar en pacientes con distintos tipos de trastornos hepatobiliares, pero esta alteración en la proporción es única del SDJ.[5][33][34] Se ha observado que los neonatos sanos suelen tener aumentos notables de los niveles coproporfirina en orina y que más del 80% corresponde al isómero I durante los primeros 2 días de vida; pero para el día 10, los niveles bajan y coinciden con los valores normales de los adultos.[33][35] Una exploración 99mTc del ácido iminodiacético hepatobiliar (gammagrafía de las vías biliares) puede ser útil si el diagnóstico no es claro o si se sospecha de otra enfermedad.[30]
Gammagrafía de las vías biliares
La recaptación del radionucleido 99mTc por parte del hígado es excelente, pero la excreción del hígado a las vías biliares es deficiente. Se obtiene una colescintigrafía única, que muestra una visualización intensa y prolongada del hígado, con una visualización retrasada de la vesícula biliar y el conducto biliar común. Este patrón es diferente del observado en los pacientes con enfermedad hepatocelular, obstrucción biliar extrahepática o síndrome de Rotor.
Biopsia hepática
Se recomienda una biopsia hepática percutánea para todos los pacientes con sospechas de padecer SDJ a fin de establecer el diagnóstico y descartar una patología hepática más grave. La histología hepática normal y la deposición parenquimatosa característica de un pigmento similar a la melanina establecerán el diagnóstico. Alternativamente, puede observarse un hígado oscuro durante una cirugía realizada por otra causa, lo que motiva la realización de una biopsia hepática. La coproporfirina en orina es un marcador sustituto y no una prueba definitiva. El SDJ es una enfermedad extremadamente infrecuente; por lo tanto, una prueba diagnóstica definitiva que confirme el diagnóstico y descarte otras enfermedades será de utilidad para tranquilizar a los pacientes con respecto a la naturaleza benigna de esta enfermedad.)
No se requiere mayor investigación, pero es posible que se hayan realizado investigaciones adicionales como parte de una evaluación general de la ictericia o antes de realizar el diagnóstico de SDJ:
En general, se suele realizar una prueba de ultrasonido del hígado y del árbol biliar; una colecistografía oral no permite visualizar la vesícula biliar, incluso si se lleva a cabo con una dosis de contraste complementaria.
Observar el hígado oscuro durante una cirugía, puede motivar una biopsia hepática para descartar una patología más grave. La histología hepática normal y la deposición parenquimatosa característica de un pigmento similar a la melanina ayudarán a establecer un diagnóstico de SDJ. Esto puede demostrarse con tinción de Fontana-Masson o a través de inmunohistoquímica. Esta llamativa pigmentación de las células hepáticas no aparece en las demás enfermedades hepáticas, incluyendo al síndrome de Rotor.[3][Figure caption and citation for the preceding image starts]: La tinción de Fontana-Masson muestra la pigmentación en un paciente con SDJColección personal del Profesor Bernard Portmann, del King's College Hospital, Londres, con autorización [Citation ends].
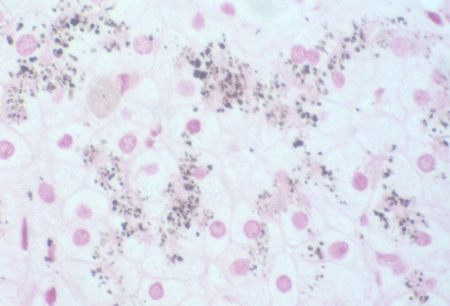 [Figure caption and citation for the preceding image starts]: Inmunohistoquímica para el transportador de aniones orgánicos multiespecífico en pacientes que muestran el pigmento pero no muestran estructuras canalicularesColección personal del Profesor Bernard Portmann, del King's College Hospital, Londres, con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Inmunohistoquímica para el transportador de aniones orgánicos multiespecífico en pacientes que muestran el pigmento pero no muestran estructuras canalicularesColección personal del Profesor Bernard Portmann, del King's College Hospital, Londres, con autorización [Citation ends].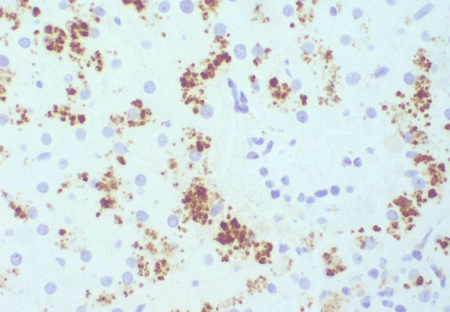 [Figure caption and citation for the preceding image starts]: Inmunohistoquímica para el transportador de aniones orgánicos multiespecífico en un control que no muestra pigmentos, pero en la que pueden observarse estructuras canaliculares con ramificación irregularColección personal del Profesor Bernard Portmann, del King's College Hospital, Londres, con autorización [Citation ends].
[Figure caption and citation for the preceding image starts]: Inmunohistoquímica para el transportador de aniones orgánicos multiespecífico en un control que no muestra pigmentos, pero en la que pueden observarse estructuras canaliculares con ramificación irregularColección personal del Profesor Bernard Portmann, del King's College Hospital, Londres, con autorización [Citation ends].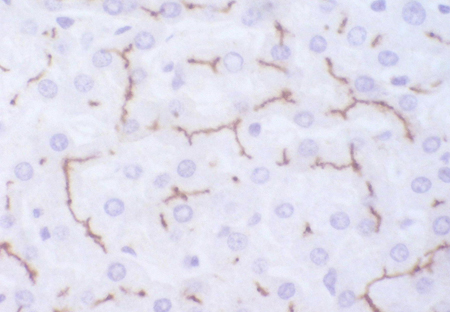
Secuenciación genética
Se ha informado el análisis mutacional del gen ABCC2 (que codifica MRP2), utilizando la secuenciación de la generación próxima y la secuenciación de Sanger como una herramienta de diagnóstico potencial.[31][36][37]
Pruebas emergentes
La proteína 2 asociada a la resistencia a múltiples fármacos (MRP2) también transporta leucotrienos a la bilis. Cuando son defectuosos, como sucede en el SDJ, se produce una mayor excreción por orina de los metabolitos de leucotrienos. Esta podría ser una prueba de diagnóstico, pero por el momento no tiene un uso clínico generalizado.[38]
Algunos estudios han informado que las mutaciones deletéreas graves que afectan a los casetes de unión al ATP de la proteína MRP2 eran más frecuentes en los pacientes con SDJ de inicio neonatal, mientras que las variantes que afectan a otros dominios de la proteína MRP2 eran más comunes en los pacientes adultos.[31][39][40]
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad