Etiology
The interaction between genetic predisposition and environmental factors is an active area of research. It is theorized that exposure of genetically susceptible individuals to certain environmental factors results in many congenital abnormalities, including orofacial clefts.
Genetic factors
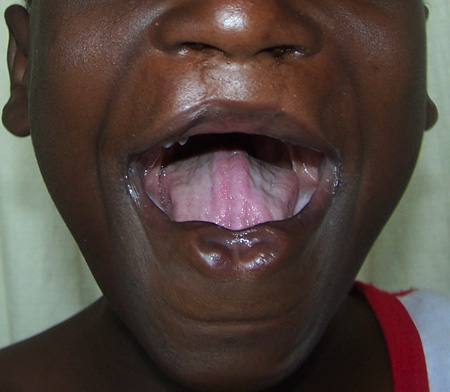
Orofacial clefts have been associated with over 300 syndromes, caused by gene mutations, teratogens, or chromosomal abnormalities. Van der Woude syndrome is the most common syndromic cause of orofacial clefting, accounting for 2% of cases.[7] It is caused by a mutation in the interferon regulatory factor 6 (IRF6) gene and is characterized by cleft lip +/- cleft palate, lip pits, skin folds, syndactyly, and oral adhesions.[11][Figure caption and citation for the preceding image starts]: Repaired bilateral cleft lip showing the lip pits of van der Woude syndromeFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].
Other syndromes associated with orofacial clefting include:[12]
Oculoauriculovertebral spectrum: this includes Goldenhar syndrome, an autosomal-dominant condition characterized by variable expression of hemifacial microsomia, ocular dermoids, renal and vertebral anomalies, and auricular deformities.
Oral-facial-digital syndrome: X-linked condition characterized by mandibular hypoplasia and cleft palate.
Treacher-Collins syndrome: autosomal-dominant condition characterized by lower lid colobomas, downward-slanting palpebral fissures, hypoplastic zygomatic arches, and low-set ears.
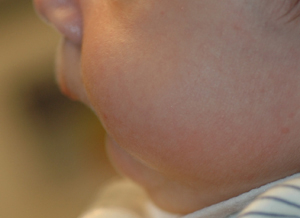
Pierre Robin sequence (or Robin sequence): classic triad of glossoptosis, microgenia, and U-shaped cleft palate caused by intrauterine deformation of the palate by a prominent tongue (pushed upward by a small jaw).[Figure caption and citation for the preceding image starts]: Lateral view of infant with Pierre Robin sequenceFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].
 [Figure caption and citation for the preceding image starts]: Infant with Pierre Robin sequence with external mandibular distraction osteogenesis device in placeFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].
[Figure caption and citation for the preceding image starts]: Infant with Pierre Robin sequence with external mandibular distraction osteogenesis device in placeFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].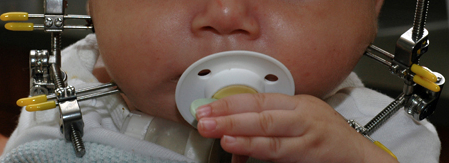
Stickler syndrome: autosomal-dominant condition associated with Pierre Robin sequence, progressive myopia, hearing loss, and arthropathy.
Velocardiofacial syndrome: autosomal-dominant condition secondary to a 22q11.2 deletion characterized by variable expression of cardiac defects, broad nasal route and skull base, and velopharyngeal dysmorphology.
Nonsyndromic familial clefts also occur. Studies show that monozygotic twins have 43% paired concordance, compared with 5% in dizygotic twins.[13] Parents without clefts have a 4% recurrence rate in future pregnancies following the birth of 1 affected child.[14] However, the risk for a third child after having 2 affected children rises from 4.4% to 9%.[15] A parent with cleft lip and/or palate has a 4% risk of having an affected child. If further first-degree relatives are affected by a cleft palate, the risk to the child rises to between 10% and 20%.[14]
Orofacial clefting shows variable severity and phenotypic expression in affected family members, and there is limited evidence to support the existence of a carrier state. First-degree relatives of patients with cleft lip and palate often demonstrate less-obvious facial abnormalities that, although difficult to prove, may act as a carrier state for this or other craniofacial disorders. Many unaffected individuals have subtle nasal asymmetries, and the parents of children with cleft lip and palate sometimes have significantly different cephalometric morphology in comparison with the noncleft population.[16][17] However, lack of consistency in the cephalometric study designs makes the exact locations of these craniofacial differences difficult to ascertain.[18] Definitive genetic studies of these families and continued anthropometric documentation of craniofacial abnormalities in relatives of affected patients are required in order to uncover previously unidentified associations. In one volumetric study of brains of boys with cleft lip, those with right-sided clefts were shown to have more abnormalities. This finding, along with other behavioral and cognitive differences found between males and females and left- and right-sided clefts, may allow the further subclassification of orofacial clefts. Possibly, laterality and sex play a role in embryologic differences between cleft types and may assist in understanding the pathophysiologic mechanisms.[19]
Environmental factors
Teratogens: anticonvulsant drugs such as phenytoin taken during pregnancy have been linked to a number of congenital deformities including cleft lip and palate.[20]
Nutritional deficiencies: conflicting reports have been published regarding the efficacy of prenatal folic acid supplementation to reduce the risk of orofacial clefting. Some studies show a decreased risk of clefting, while others show no reduction.[21][22] A Cochrane review of randomized controlled trials showed that prenatal folic acid had an overall protective effect against neural tube defects. However no relation to decreased cleft lip and palate was found.[23] Folic acid, alone or in combination with vitamins and minerals, prevents neural tube defects but does not have a clear effect on other birth defects.
Maternal perinatal multivitamin use has been shown to be more protective against cleft lip and palate in infants with certain transforming growth factor-alpha (TGF-A) genotypes.[24] It has been shown that recurrence rates for clefting (i.e., the incidence of clefting if one sibling has already been born with an orofacial cleft) is decreased significantly with the use of folic acid, although no dose-dependent relationship has been established.[25]
Maternal tobacco use: although the pathophysiology behind the association is not yet understood, the risk of orofacial clefting is increased in mothers who use tobacco during pregnancy or periconceptionally (from 1 month before to 3 months after conception). There are a number of meta-analyses that show a statistically significant, dose-dependent link between maternal smoking and cleft lip with or without cleft palate and isolated cleft palate.[26][27][28][29][30]
Maternal alcohol consumption: higher quantities of maternal alcohol consumption (>5 units per drinking session) are associated with an increased incidence of orofacial clefts in women with particular alcohol dehydrogenase gene variants.[31]
Pathophysiology
The embryologic development of the upper lip and nose requires a sequence of complex, genetically programmed events. This involves fusion of the 5 major facial prominences, occurring between the 3rd and 8th week of gestation, with lip development between the 3rd and 7th weeks, and palate development between the 5th and 12th weeks. [Figure caption and citation for the preceding image starts]: Diagram of the 5-week-old embryo with the 5 major facial prominences labeled (the medial and lateral nasal prominences develop from the frontonasal prominence)Original drawing by Dr Amir Rafii [Citation ends].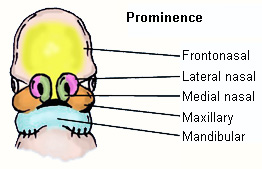
The complexity of this craniofacial developmental pathway and the numerous developmental points at which clefting could be induced is reflected in the heterogeneity of the phenotypic expression of the condition.
Cleft lip and/or palate: the maxillary, medial nasal, and lateral nasal prominences converge through a complicated process of epithelial bridging, programmed cell death, and subepithelial-mesenchymal penetration.[32][33] Cleft lip and/or palate is likely to be secondary to a defect of epithelial fusion or mesenchymal growth, processes involving many possible genetic loci or intracellular signaling pathways.[34] This results in interrupted fusion of the maxillary and median nasal prominences. In bilateral cleft lip with or without cleft palate, the arterial network and musculature of the lateral elements parallel that of the lateral segment of the unilateral deformity. The abnormal insertion of the cleft lip musculature follows the cleft margin up to the piriform aperture, and the prolabial segment receives its blood supply from the septal, columellar, and premaxillary vessels.
Isolated unilateral cleft lip: the orbicularis oris (OO) is a ring of concentric muscle that constricts and puckers the sphincter of the mouth. In isolated unilateral cleft lip, the OO fibers on the cleft side insert into the nasal base, and the central (noncleft) OO fibers abnormally insert into the nasal spine and septum. This causes the base of the nose to splay laterally when the infant smiles.
Isolated cleft palate: the development of the palate involves fusion of the lateral palatal shelves and nasal septum in an anteroposterior direction from the incisive foramen to the uvula. A cleft palate is formed when normal palatal development is interrupted before the 12th week of gestation. The degree of clefting can range from a complete isolated cleft palate to a bifid uvula. Deformational cleft palate is seen in Pierre Robin sequence, where a small mandible (micrognathia) limits the space for the tongue, and the prominent tongue (glossoptosis) mechanically obstructs palatal fusion, leading to the classic triad of micrognathia, glossoptosis, and an isolated cleft palate.
Midline clefts of the nose and/or lip: these are likely to arise from an interruption in the fusion of the paired median nasal prominences during embryologic development. Most median facial deformities represent developmental field defects and are sporadic, with multiple etiologic factors.
Classification
Veau classification[1]
Classification system proposed in 1938.
Group I (A)
Defects of the soft palate alone
Group II (B)
Defects involving the hard and soft palates (not extending anterior to the incisive foramen)
Group III (C)
Defects involving the palate through to the alveolus
Group IV (D)
Complete bilateral clefts.
Kernahan and Stark classification[2]
Embryology-based classification system proposed in 1958 that designates the incisive foramen as the dividing line between the primary and secondary palates.
The incisive foramen is a funnel-shaped opening through which neurovascular bundles pass. It is located in the hard palate behind the middle upper teeth (incisors). This structure is an important embryologic landmark, which is used to define the boundary between the primary and secondary palate.
Primary palate includes those structures anterior to the incisive foramen (lip, premaxilla, anterior septum).
Secondary palate includes those structures posterior to the incisive foramen (lateral palatine shelves, soft palate, and uvula).
Kernahan classification[3]
Classification system based on the resemblance of an intra-oral view of a cleft lip and palate to the letter "Y," proposed in 1971.
The area affected by the cleft is marked on the "Y" and labeled from 1 to 9, each of which represents a different anatomic structure. Combinations of the numeric values represent the appearance of the cleft lip, alveolus, or palate. [Figure caption and citation for the preceding image starts]: Kernahan striped "Y" cleft classificationFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].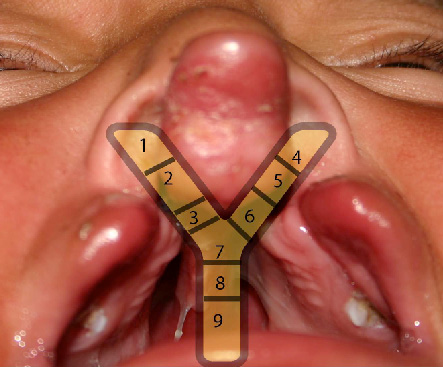
Areas 1 and 4 represent the right and left side of the nasal floor, respectively.
Areas 2 and 5 represent the right and left side of the lip, respectively.
Areas 3 and 6 represent the right and left side of the paired alveolar segment, respectively.
Area 7 represents the primary palate.
Areas 8 and 9 represent the secondary palate.
Harkins classification[4]
Classification system proposed in 1962.
Cleft of primary palate
Cleft lip
Alveolar cleft
Cleft of secondary palate
Soft palate
Hard palate
Mandibular process clefts
Naso-ocular clefts: involving the nose toward the medial canthal region
Oro-ocular clefts: extending from the oral commissure toward the palpebral fissure
Oro-aural clefts: extending from the oral commissure toward the auricle.
Spina classification[5]
Classification system proposed in 1974.
Preincisive foramen clefts (lip ± alveolus)
Unilateral
Bilateral
Median
Transincisive foramen cleft (lip, alveolus, palate)
Unilateral
Bilateral
Postincisive foramen clefts (secondary cleft palate)
Atypical (rare) facial clefts.
Tessier classification[6]
Tessier described a classification scheme that is universally utilized, in a landmark article of 1976. [Figure caption and citation for the preceding image starts]: Tessier craniofacial cleft classificationFrom: Tessier P. Anatomical classification of facial, cranio-facial, and latero-facial clefts. J Maxillofac Surg. 1976;4:69-92 [Citation ends].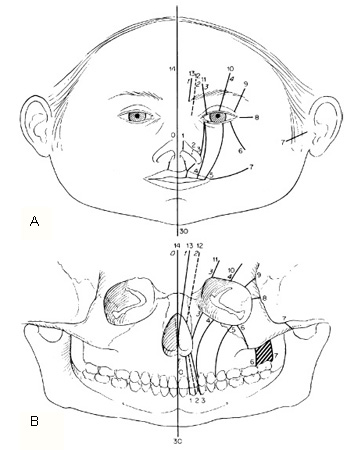 [Figure caption and citation for the preceding image starts]: Infant with right incomplete Tessier No. 3 atypical craniofacial cleftFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].
[Figure caption and citation for the preceding image starts]: Infant with right incomplete Tessier No. 3 atypical craniofacial cleftFrom the collection of Travis T. Tollefson, MD, FACS [Citation ends].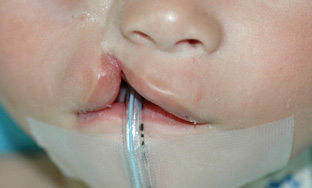
Orofacial clefts can manifest as:
Unilateral or bilateral
Complete, incomplete, or microform (e.g., submucous cleft palate)
Clefting of the lip with or without the palate, or of the palate in isolation
Atypical craniofacial clefts.
Use of this content is subject to our disclaimer