Humans and some other higher primates develop gout spontaneously. Humans no longer express the gene for the enzyme uricase, which, in animals, degrades uric acid to the more soluble compound allantoin. This, coupled with a high rate of renal reabsorption of urate, results in hyperuricemia and gout.[19]Johnson RJ, Rideout BA. Uric acid and diet - insights into the epidemic of cardiovascular disease. N Engl J Med. 2004;350:1071-1073.
http://www.ncbi.nlm.nih.gov/pubmed/15014177?tool=bestpractice.com
[20]Wu XW, Lee CC, Muzny DM, et al. Urate oxidase: primary structure and evolutionary implications. Proc Natl Acad Sci USA. 1989;86:9412-9416.
https://www.pnas.org/content/pnas/86/23/9412.full.pdf
http://www.ncbi.nlm.nih.gov/pubmed/2594778?tool=bestpractice.com
[21]Wu XW, Muzny DM, Lee CC, et al. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J Mol Evol. 1992;34:78-84.
http://www.ncbi.nlm.nih.gov/pubmed/1556746?tool=bestpractice.com
[22]Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016 Oct 22;388(10055):2039-52.
http://www.ncbi.nlm.nih.gov/pubmed/27112094?tool=bestpractice.com
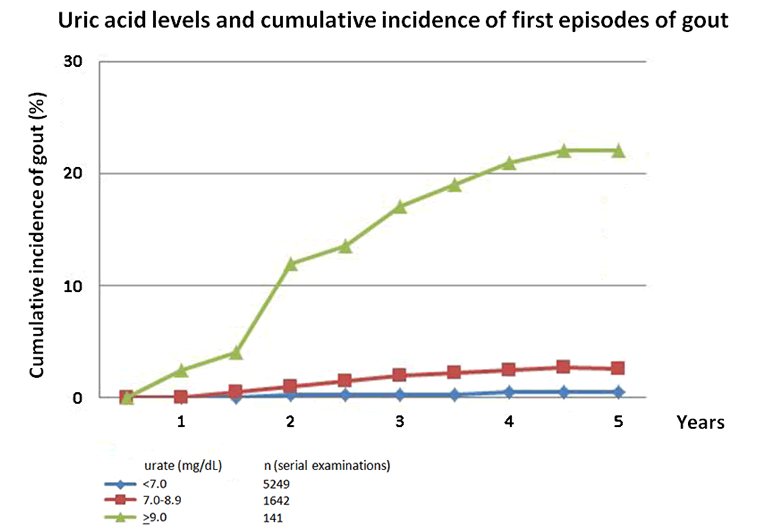
Uric acid exists as urate at a physiologic pH. High urate levels result in supersaturation and crystal formation, leading to gout. Urate levels directly correlate with risk for gout.[9]Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med. 1987;82:421-426.
http://www.ncbi.nlm.nih.gov/pubmed/3826098?tool=bestpractice.com
Drugs that reduce urate levels decrease the risk of recurrent attacks.[23]Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51:321-325.
https://onlinelibrary.wiley.com/doi/full/10.1002/art.20405
http://www.ncbi.nlm.nih.gov/pubmed/15188314?tool=bestpractice.com
The solubility of urate in the joints depends on temperature, pH, nonaggregated proteoglycans, and other factors. Gout more commonly affects the first metatarsophalangeal joint (cool part of the body) and osteoarthritic joints.[24]Fam AG, Stein J, Rubenstein J. Gouty arthritis in nodal osteoarthritis. J Rheumatol. 1996;23:684-689.
http://www.ncbi.nlm.nih.gov/pubmed/8730127?tool=bestpractice.com
Urate crystals in the joint interact with undifferentiated phagocytes and trigger an acute inflammatory response by inducing tumor necrosis factor (TNF)-alpha and activating signal pathways and endothelial cells.[25]Yagnik DR, Hillyer P, Marshall D, et al. Noninflammatory phagocytosis of monosodium urate monohydrate crystals by mouse macrophages. Implications for the control of joint inflammation in gout. Arthritis Rheum. 2000;43:1779-1789.
https://onlinelibrary.wiley.com/doi/epdf/10.1002/1529-0131%28200008%2943%3A8%3C1779%3A%3AAID-ANR14%3E3.0.CO%3B2-2
http://www.ncbi.nlm.nih.gov/pubmed/10943868?tool=bestpractice.com
TNF-alpha, interleukin (IL)-8, and other chemokines lead to neutrophil adhesion to endothelium, influx, and amplification, resulting in neutrophilic synovitis.
Colchicine works by intercepting the neutrophil-endothelial interaction.[10]Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499-516.
http://www.ncbi.nlm.nih.gov/pubmed/16204163?tool=bestpractice.com
[26]Cronstein BN, Molad Y, Reibman J, et al. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest. 1995 Aug;96(2):994-1002.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC185287
http://www.ncbi.nlm.nih.gov/pubmed/7543498?tool=bestpractice.com
IL-8 accounts for 90% of the chemotactic activity of neutrophils in response to uric acid crystals; hence, inhibiting IL-8 may have therapeutic implications.[27]Terkeltaub R, Zachariae C, Santoro D, et al. Monocyte-derived neutrophil chemotactic factor/interleukin-8 is a potential mediator of crystal-induced inflammation. Arthritis Rheum. 1991;34:894-903.
http://www.ncbi.nlm.nih.gov/pubmed/2059236?tool=bestpractice.com
[28]Nishimura A, Akahoshi T, Takahashi M, et al. Attenuation of monosodium urate crystal-induced arthritis in rabbits by a neutralizing antibody against interleukin-8. J Leukoc Biol. 1997;62:444-449.
http://www.ncbi.nlm.nih.gov/pubmed/9335313?tool=bestpractice.com
In addition there is evidence that urate crystals activate NALP3 inflammasome (a key regulator of IL-1beta secretion), which plays a role in the gout inflammatory reaction.[29]Martinon F, Pétrilli V, Mayor A, et al. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-241.
http://www.ncbi.nlm.nih.gov/pubmed/16407889?tool=bestpractice.com
Urate crystals can induce chondrocytes to produce metalloproteinase and nitric oxide, and chronic inflammation, leading to synovitis, cartilage loss, bone damage by inhibiting osteoblasts, and bone erosions.[30]Liu R, Liote F, Rose DM, et al. Proline-rich tyrosine kinase 2 and Src kinase signaling transduce monosodium urate crystal-induced nitric oxide production and matrix metalloproteinase 3 expression in chondrocytes. Arthritis Rheum. 2004;50:247-258.
https://onlinelibrary.wiley.com/doi/full/10.1002/art.11486
http://www.ncbi.nlm.nih.gov/pubmed/14730623?tool=bestpractice.com
[31]Bouchard L, de Medicis R, Lussier A, et al. Inflammatory microcrystals alter the functional phenotype of human osteoblast-like cells in vitro: synergism with IL-1 to overexpress cyclooxygenase-2. J Immunol. 2002;168:5310-5317.
https://www.jimmunol.org/content/168/10/5310.full
http://www.ncbi.nlm.nih.gov/pubmed/11994489?tool=bestpractice.com
Genetic mutations that potentially predispose people to hyperuricemia and gout have been identified.[32]Akashi A, Nakayama A, Kamatani Y, et al. A common variant of LDL receptor related protein 2 (LRP2) gene is associated with gout susceptibility: a meta-analysis in a Japanese population. Hum Cell. 2020 Apr;33(2):303-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7080676
http://www.ncbi.nlm.nih.gov/pubmed/31975031?tool=bestpractice.com
[33]Zhou Z, Li Z, Wang C, et al. Common variants in the SLC28A2 gene are associated with serum uric acid level and hyperuricemia and gout in Han Chinese. Hereditas. 2019 Jan 16;156:4.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6335706
http://www.ncbi.nlm.nih.gov/pubmed/30679935?tool=bestpractice.com
[34]Nakayama A, Nakatochi M, Kawamura Y, et al. Subtype-specific gout susceptibility loci and enrichment of selection pressure on ABCG2 and ALDH2 identified by subtype genome-wide meta-analyses of clinically defined gout patients. Ann Rheum Dis. 2020 May;79(5):657-65.
https://ard.bmj.com/content/79/5/657.long
http://www.ncbi.nlm.nih.gov/pubmed/32238385?tool=bestpractice.com
[35]Kawaguchi M, Nakayama A, Aoyagi Y, et al. Both variants of A1CF and BAZ1B genes are associated with gout susceptibility: a replication study and meta-analysis in a Japanese population. Hum Cell. 2021 Mar;34(2):293-9.
https://www.doi.org/10.1007/s13577-021-00485-4
http://www.ncbi.nlm.nih.gov/pubmed/33517564?tool=bestpractice.com
[36]Lukkunaprasit T, Rattanasiri S, Turongkaravee S, et al. The association between genetic polymorphisms in ABCG2 and SLC2A9 and urate: an updated systematic review and meta-analysis. BMC Med Genet. 2020 Oct 21;21(1):210.
https://www.doi.org/10.1186/s12881-020-01147-2
http://www.ncbi.nlm.nih.gov/pubmed/33087043?tool=bestpractice.com
However, further study is required to determine their clinical significance.
Spontaneous resolution of gout attack results from clearance of urate crystals by differentiated phagocytes, coating of the crystals with proteins, neutrophilic apoptosis, and inactivation of inflammatory mediators.[10]Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499-516.
http://www.ncbi.nlm.nih.gov/pubmed/16204163?tool=bestpractice.com