Summary
Definition
Noonan syndrome (NS) is a relatively common, autosomal-dominant, inherited disorder that is predominantly characterized by short stature, subtle facial dysmorphisms, chest deformity, congenital heart disease, and variable degrees of developmental delay.[1] Coagulation defects, cryptorchidism in men, and lymphatic dysplasia are not uncommon.[Figure caption and citation for the preceding image starts]: Boy (6 years of age) with Noonan syndromeFrom the collection of Judith E. Allanson [Citation ends].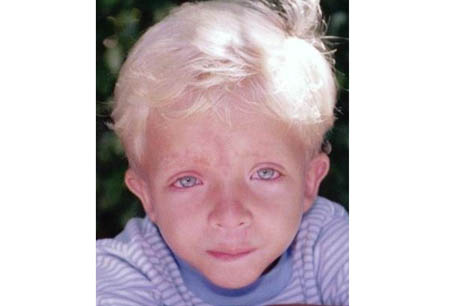
The syndrome is thought to be caused primarily by gain-of-function (activating) mutations in genes in the Ras/mitogen-activated protein kinase (MAPK) signal transduction pathway.[2][3][4][5][6][7][8][9][10] Genes implicated include: PTPN11, SOS1, RAF1, RIT1, RASA2, LZTR1, SOS2, CBL, KRAS, NRAS, BRAF, PPP1CB, and MAP2K1. The most commonly identified genetic mutation involves PTPN11.[2][11][12]
Noonan syndrome caused by SHOC2 mutation is now considered by most to be an overlapping condition with unusual features (loose anagen hair); some consider it to be a distinct condition, while others consider it to be classified only as Noonan syndrome.
History and exam
Key diagnostic factors
- positive family history
- short stature
- dysmorphic facial features
- cryptorchidism
- cardiac anomalies
- delayed puberty
- easy bruising or bleeding
- lymphedema
- pigmentary anomalies
- sparse or absent eyebrows and lashes
- splenomegaly
Other diagnostic factors
- abnormalities identified prenatally
- chest deformity
- developmental delay/learning difficulty
- skeletal anomalies
- muscle weakness
- history of renal malformation
Diagnostic tests
Tests to consider
- CBC
- coagulation profile
- molecular genetic testing
- abdominal ultrasound
- renal ultrasound
Treatment algorithm
Contributors
Authors
David A. Stevenson, MD
Associate Professor
Department of Pediatrics
Division of Medical Genetics
Stanford University
Stanford
CA
Disclosures
DAS has acted as a consultant for Lineagen, GLG, and Alexion, and has given expert testimony. He is on the medical advisory board for parents' support groups for Costello syndrome and CFC syndrome. DAS is an author of a reference cited in this topic. DAS has also been reimbursed by RASopathies Network for attending conferences.
Acknowledgements
Dr David A. Stevenson would like to gratefully acknowledge Dr Judith E. Allanson, the previous contributor to this topic. JEA is an author of a number of references cited in this topic.
Peer reviewers
Liliana N. Contreras, MD
Chief
Endocrine Research Department
Instituto de Investigaciones Médicas Alfredo Lanari
IDIM-CONICET
University of Buenos Aires
Argentina
Disclosures
LNC declares that she has no competing interests.
Bruce Gelb, MD
Professor of Pediatrics
Mount Sinai School of Medicine
New York
NY
Disclosures
BG received royalties from GeneDx, Correlegan, Preventative Genetics, Baylor College of Medicine, and Harvard Medical School/Partners for genetic testing of Noonan syndrome. BG is an author of a number of references cited in this topic.
Jacqueline Noonan, MD
Professor Emeriti
Department of Pediatrics
College of Medicine
University of Kentucky
Lexington
KY
Disclosures
JN is on the Noonan Syndrome Advisory Board for Novo Nordisk and has received payment for speaking at a symposium. JN is an author of a number of references cited in this topic.
Use of this content is subject to our disclaimer