Approach
HS can typically be diagnosed easily at any age. It is most commonly diagnosed as a mild condition in children, where there may be jaundice (variable in severity over time) and splenomegaly. Routine laboratory tests usually confirm the diagnosis by detection of:
Increased red-cell turnover (reticulocytosis) ± anemia
Typical spherocytes on the blood smear
Associated absence of an immune cause (negative direct antiglobulin test [DAT]).
Confirmatory laboratory tests may be required in people with atypical features.[15] Rarely, additional specialized tests are required.
When a diagnosis of HS is suspected, patients should be referred to a hematology specialist for definitive diagnosis and routine monitoring.
[Figure caption and citation for the preceding image starts]: Diagnostic algorithm for HSCreated by the BMJ Evidence Centre using information from author [Citation ends].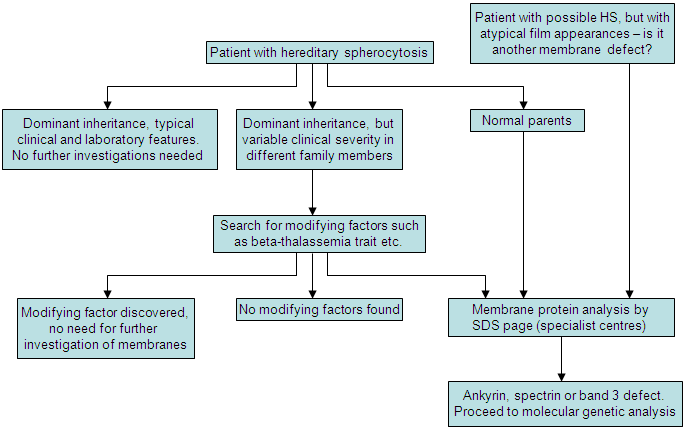
History
HS is the most common cause of hemolysis in white people of Northern European ancestry. HS may be found in most racial groups, but is less common in black people.[16] Most people with HS will have a family history of anemia, jaundice, splenectomy, or known HS.
Presentation may be at any age. More severely affected individuals tend to present earlier in life. Clinical severity ranges from no symptoms, to fatigue from severe anemia, and jaundice. Often, when there are no symptoms, the condition is detected as an incidental finding when a blood test is performed for another reason.
Children or adults may be asymptomatic until they contract parvovirus B19 infection with resultant aplastic crisis.[17] This virus directly attacks erythroid precursors in the bone marrow and results in erythroid aplasia for about 10 days. Patients with a shortened red-cell life span, such as occurs in HS (and other hemolytic conditions), have a rapidly progressive anemia during this period of absent erythropoiesis and typically present with acute onset of marked pallor, lethargy, and fever. The hemoglobin (Hb) value is usually between 3 and 6 g/dL. Reticulocyte count is typically <1%. During the recovery phase the reticulocyte count increases, and following recovery from an aplastic crisis, permanent immunity usually results. Parvovirus B19 infection may unmask hitherto undiagnosed HS in a family.
Symptoms and signs may vary over time. Hyperhemolytic crises are more common but less severe than aplastic episodes, and are characterized by acceleration of the usual hemolytic process with resulting exacerbation of symptoms. These episodes usually accompany nonspecific viral infections, in which the reticuloendothelial system undergoes hyperplasia with further enlargement of the spleen. The episodes may be repetitive.[Figure caption and citation for the preceding image starts]: Presentation of HS by ageCreated by the BMJ Evidence Centre using information from author [Citation ends].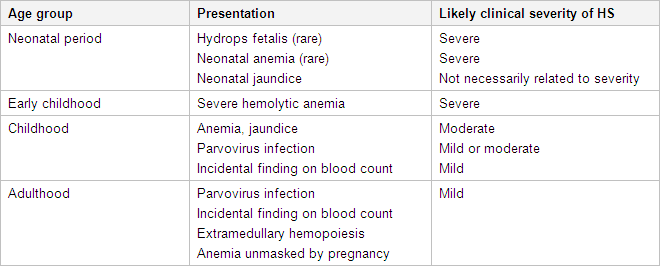
Physical exam
Many individuals with HS have mild jaundice. It is a common feature when HS presents in the neonatal period, where severity of the jaundice is not necessarily related to future severity of HS. In some cases, the jaundice is severe enough to warrant exchange transfusion.[5] Very rarely, HS may present with hydrops fetalis or stillbirth due to severe anemia (when an infant inherits defects in membrane proteins from both parents).[9][10][11]
The diagnosis of HS should be considered in any individual with unexpected or unexplained splenomegaly at any age. Splenomegaly (mild to moderate enlargement) is very common in HS of all severities but is not specific to this disorder.[5] It is caused by the trapping and destruction of the abnormal, inflexible red blood cells in the spleen. The splenomegaly generally does not have any symptoms or clinical consequences. However, during hyperhemolytic crises the spleen rarely may become acutely and significantly enlarged and cause left upper quadrant abdominal pain and symptoms of early satiety. The spleen generally shrinks to its previous size between episodes.
Individuals with HS may or may not have signs of anemia. Depending on the severity of the condition, signs of anemia will be variable: from no obvious signs to severe pallor.[Figure caption and citation for the preceding image starts]: Jaundiced sclera in eye of child with HSFrom the collection of Paula Bolton-Maggs, University of Manchester, UK; used with permission [Citation ends].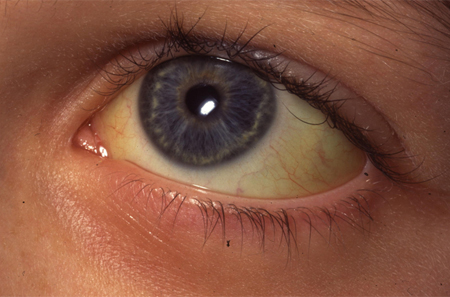
Initial tests
Many cases are detected as an incidental finding when a blood count is done for another purpose. The diagnosis should also be considered in any patient with jaundice or splenomegaly, or with family history of HS in a first-degree relative.[18]
The diagnosis should be considered in the context of an unexpectedly low Hb result at any age.
Complete blood count and smear
In most cases a blood count and smear is diagnostic. The patient has a normal or reduced Hb, normal or reduced MCV, and often an elevated MCHC. Abnormal red-cell morphology of spherocytes is present on the blood smear, with increased reticulocytes. The blood smear may demonstrate pincer cells (mushroom-shaped cells) in addition to spherocytes, which are due to band 3 protein mutations. Anemia may be absent in HS when the marrow output increases sufficiently to keep the Hb normal, but there will be an increase in reticulocytes (known as compensated hemolysis).
Neonatal blood smears can be difficult to interpret. Careful review of the blood smear is needed with follow-up if the diagnosis is not clear. Typical blood smear appearances of spherocytes are easier to see after a few months.
Differentials
Careful analysis of the red-cell morphology from the blood smear is very important in order not to miss alternative less-common disorders.[16] The presence of spherocytes is not limited to HS and other diagnoses must be ruled out by appropriate history, the clinical setting, and investigations.[16]
The most important alternative diagnosis for spherocytes is autoimmune hemolytic anemia (AIHA). In AIHA, abnormal antibodies coat the red cells and can be detected by the direct antiglobulin test (DAT), which is negative in HS. In infancy, hemolysis caused by irregular maternal IgG antibodies must also be excluded as a possible cause.
Differentiation of HS from other inherited red-cell membrane disorders is important. Many of these are obvious from the blood smear appearances (e.g., elliptocytosis), but where the red-cell appearances are atypical, other diagnoses should be considered. It is particularly important to rule out hereditary stomatocytosis with membrane transport defects, because splenectomy carries a particularly high risk of venous thromboembolism postoperatively. Serum bilirubin and liver aminotransferases are performed if jaundice is suspected clinically. Cases of congenital dyserythropoietic anemia have also been misdiagnosed as HS.[Figure caption and citation for the preceding image starts]: Blood smear of patient with HS (A) compared with normal blood smear (B); Pincer cell (mushroom-shaped cell) indicatedFrom the collection of Paula Bolton-Maggs, University of Manchester, UK; used with permission [Citation ends].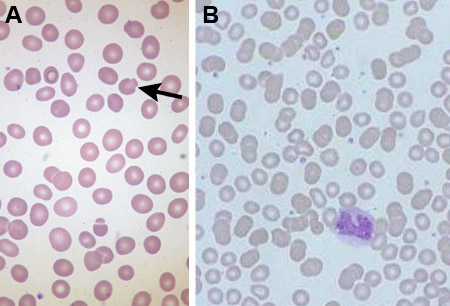 [Figure caption and citation for the preceding image starts]: Blood smear of a patient with HS; spherocyte indicatedFrom the collection of Shelley Crary, University of Texas Southwestern Medical Center, TX; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Blood smear of a patient with HS; spherocyte indicatedFrom the collection of Shelley Crary, University of Texas Southwestern Medical Center, TX; used with permission [Citation ends].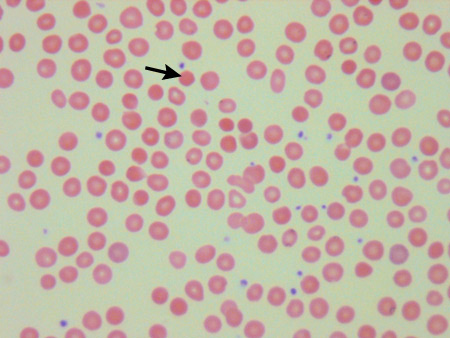
Confirmatory tests to consider
The typical case of HS does not require any confirmatory test. If the diagnosis is equivocal, a confirmatory test may be considered.
Eosin-5-maleimide (EMA) binding test
Confirmation using the eosin-5-maleimide binding test is recommended when the features are not typical (e.g., the morphology on the blood smear is not quite typical or there is no family history).[15] The EMA binding test is based on fluorescent intensity measured by flow cytometry.[19][20][21]
The EMA binds to band 3 protein, which is disrupted in HS resulting in decreased fluorescence. The EMA binding test has been reported to have a sensitivity of 92.7% for HS and a specificity of 99.1%, but may be abnormal in other red-cell disorders (particularly congenital dyserythropoietic anemia type II (CDA-II).[19]
Acidified glycerol lysis test
Uses 20 microliters of whole blood and measures the time taken for the absorbance to fall to one half of its original value using a red-cell suspension before and after addition of glycerol. This test has a reported sensitivity of 98.3% for HS and a specificity of 91.1%, but is also positive in AIHA, pregnancy, myelodysplasia, and some other conditions.[22] The acidified glycerol lysis test is not widely available.
Additional tests in difficult cases
The typical case of HS does not require additional tests. Additional tests are indicated:
When the clinical phenotype is more severe than expected from the red-cell appearances
When the red-cell abnormalities are more severe than seen in the one known affected parent
Where splenectomy is considered and the morphology is atypical.
Quantitative protein analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) may be undertaken in atypical cases (but is not widely available). SDS-PAGE analyzes red-cell membrane content and establishes which membrane protein is deficient by demonstrating protein bands for spectrin, ankyrin, band 3, and protein 4.2 (the membrane proteins that can be affected in HS).[15] A survey of 300 patients with HS in Italy showed that band 3 and spectrin deficiencies were the most common protein abnormalities.[3]
It is important to exclude rare forms of red-cell disorder where splenectomy is contraindicated.[5] Splenectomy is of little value in congenital dyserythropoietic anemia type II (CDA-II), which may be confused with HS.
Genetic analysis
Mutations have been described in 5 cytoskeletal proteins (alpha- and beta-spectrin, ankyrin, band 3, and protein 4.2).[4] [23]
Most cases of dominant HS are caused by mutations in genes for ankyrin, band 3, or beta-spectrin. Mutations in these genes may cause a secondary defect of other skeletal proteins. Mutations in the gene for protein 4.2 are more common in Japan. Nondominant HS can occur as a result of inheritance of a pathogenetic mutation from one parent and a silent low expressed allele from the other, or from de novo mutations.
If there are existing genetic test results, do not perform repeat testing unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[24]
Use of this content is subject to our disclaimer