Approach
Retinoblastoma most commonly presents with leukocoria (white pupillary reflex), strabismus, or orbital pseudocellulitis.[8] It often causes an exudative retinal detachment with vitreous seeding. Ophthalmic ultrasound typically shows variable or high internal reflectivity with foci of calcification. The diagnosis is made without a biopsy.
If retinoblastoma is suspected, it is essential that a proper assessment is made by an appropriate team of specialists, led by an ocular oncologist. Early diagnosis is critical because prognosis is associated with disease extent.[20] Access to subspecialized health care has also been associated with outcomes.[42]
History
Although most retinoblastomas present in children under 3 years of age, cases have been reported in children aged 7 years or older.[3] Retinoblastoma can sometimes be detected in utero using ultrasound or may be identified shortly after birth, particularly if there is a known family history of the condition (approximately 10% of patients).
Children born to these families are examined for the disease shortly after birth and monitored closely thereafter. In the remaining 90% of cases, the diagnosis may be suspected when certain features are noted by parents or other caregivers.[23][25]
Leukocoria is the most common sign of retinoblastoma and can be seen in approximately 60% of cases.[8] It is often noted in photographs of children taken with a flash. Less commonly, strabismus or orbital pseudocellulitis may be seen.[9][12] Retinoblastoma can be unilateral or bilateral.
Other presentations
Include decreased vision, glaucoma, hypopyon, and ocular pain.[11][13] Occasionally, bilateral retinoblastomas occur with a concomitant pineal primitive neuroectodermal tumor (pinealoma), a condition termed trilateral retinoblastoma.
Children with 13q syndrome may present with features of retinoblastoma in addition to other characteristic systemic features, such as: mental and growth retardation; craniofacial dysmorphisms; hand and foot anomalies; and defects of the brain, heart, and kidneys.[15] Rarely, retinoblastoma presents in conjunction with other pediatric ophthalmic conditions such as morning glory syndrome (a congenital defect in which there is a cleft in the optic disc) or retinopathy of prematurity.
In developed countries, the percentage of patients presenting with metastatic disease is extremely small.[9] Common sites of metastases include the optic nerve/choroid, orbit, central nervous system (CNS), bone marrow, bone, and soft tissue. Therefore, signs and symptoms associated with metastatic disease may be present.
Clinical examination and funduscopy
All children with suspected retinoblastoma should undergo a thorough bilateral eye examination. Strabismus or pseudocellulitis/edema may sometimes be noted. Leukocoria is often seen, especially with advanced retinoblastoma.[8][Figure caption and citation for the preceding image starts]: Leukocoria (white pupillary light reflex) in the left eye of a patient with unilateral retinoblastomaPersonal collection of Dr Timothy Murray [Citation ends].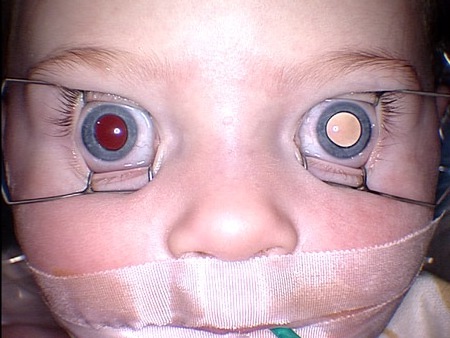
Examination under general anesthesia (EUA) should also be conducted.[43] During this examination, all critical ocular imaging should be performed and recorded (to map out the size, shape and location of any tumor).
Funduscopy reveals a characteristic chalky, white-gray retinal mass. Dilated fundus examination with 360-degree scleral depression is important to enable identification of peripheral tumors. Often there is total retinal detachment and the vessels within the detached retina can be seen behind the lens. Vitreous and subretinal seeding are frequently present.
[Figure caption and citation for the preceding image starts]: Large retinoblastoma focus in the left eyePersonal collection of Dr Timothy Murray [Citation ends].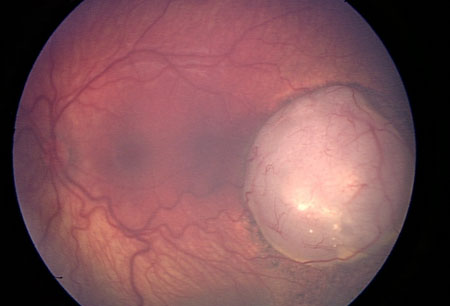 [Figure caption and citation for the preceding image starts]: Macular retinoblastoma in the right eyePersonal collection of Dr Timothy Murray [Citation ends].
[Figure caption and citation for the preceding image starts]: Macular retinoblastoma in the right eyePersonal collection of Dr Timothy Murray [Citation ends].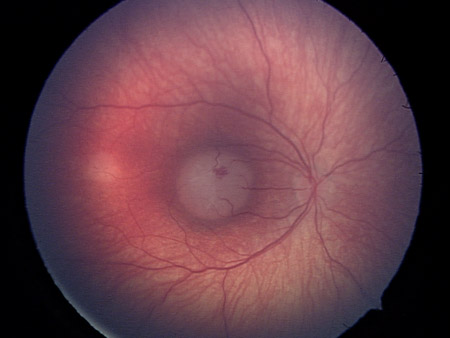 [Figure caption and citation for the preceding image starts]: Two large retinoblastoma foci in the left eye; note the associated subretinal seedingPersonal collection of Dr Timothy Murray [Citation ends].
[Figure caption and citation for the preceding image starts]: Two large retinoblastoma foci in the left eye; note the associated subretinal seedingPersonal collection of Dr Timothy Murray [Citation ends].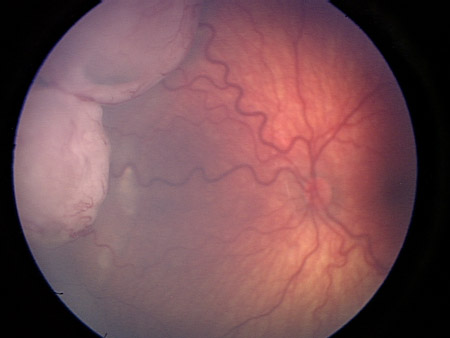 [Figure caption and citation for the preceding image starts]: Vitreous seeding associated with retinoblastomaPersonal collection of Dr Timothy Murray [Citation ends].
[Figure caption and citation for the preceding image starts]: Vitreous seeding associated with retinoblastomaPersonal collection of Dr Timothy Murray [Citation ends].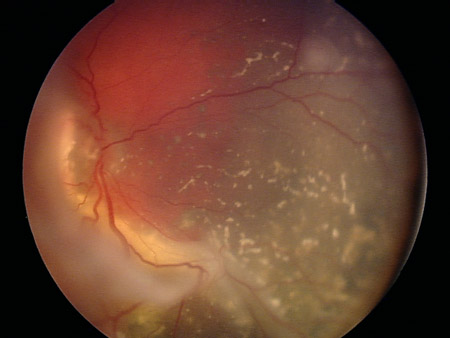
Wide-field fundus photography allows for the acquisition of high-quality posterior and peripheral retinal images and for fluorescein angiography as needed to differentiate retinoblastoma from other disease entities.[44]
Spectral domain optical coherence tomography establishes anatomic stability of the macula and fovea. Images document small areas of tumor growth, subtle vitreous seeding, and subretinal seeds, as well as frank and subtle subretinal fluid.[44]
Ophthalmic ultrasound
Standard ophthalmic A- and B-scan ultrasound should be performed to aid diagnosis.[45] It is typically performed during the initial clinical examination. If necessary, it may be carried out as part of the examination under anesthesia. With retinoblastoma, A-scan reveals variable or high internal reflectivity and B-scan typically reveals a mass filling the globe with calcification and accompanying shadowing.
[Figure caption and citation for the preceding image starts]: Ultrasound of retinoblastomaAerts I, et al. Orphanet J Rare Dis 2006 Aug 25; 1: 31; licensed under CC BY 2.0 [Citation ends].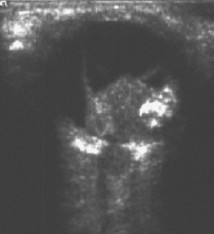
Magnetic resonance imaging (MRI) head/orbit
In general, imaging studies of the head and orbit are not necessary for the diagnosis of retinoblastoma, but are routinely used to eliminate concerns for CNS/orbital involvement.[46]
If scans are performed, retinoblastoma usually appears on MRI as an intraocular mass containing calcium. On T2-weighted MRI, retinoblastoma typically appears hypointense to vitreous.[47]
Patients diagnosed with bilateral retinoblastomas should have an MRI of the brain to exclude the possible presence of a concomitant primitive neuroectodermal tumor in the pineal gland. Although not routinely recommended, MRI of the brain may be ordered if metastatic disease is suspected.
Computed tomography (CT) scans are not recommended.[45] CT results in radiation exposure that increases the risk of secondary cancers in retinoblastoma patients with a germline mutation.[45]
[Figure caption and citation for the preceding image starts]: MRI pattern of retinoblastoma with optic nerve involvement (sagittal enhanced T1-weighted sequence)Aerts I, et al. Orphanet J Rare Dis 2006 Aug 25; 1: 31; licensed under CC BY 2.0 [Citation ends].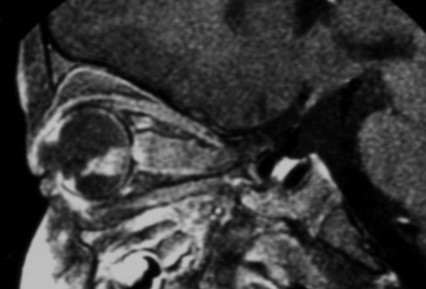 [Figure caption and citation for the preceding image starts]: Aspect of trilateral retinoblastoma (MRI)Aerts I, et al. Orphanet J Rare Dis 2006 Aug 25; 1: 31; licensed under CC BY 2.0 [Citation ends].
[Figure caption and citation for the preceding image starts]: Aspect of trilateral retinoblastoma (MRI)Aerts I, et al. Orphanet J Rare Dis 2006 Aug 25; 1: 31; licensed under CC BY 2.0 [Citation ends].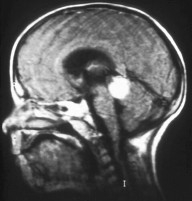
Molecular genetic testing
Molecular testing for a mutation in the RB1 gene is most useful when tumor tissue is available and/or if there are multiple family members affected. In general, results of molecular testing do not guide ocular therapy. However, the presence of a germinal mutation can be helpful for family planning and screening for secondary cancers.[14]
Bone marrow aspiration and lumbar puncture
Patients do not typically undergo testing for metastatic disease unless the suspicion is high: for example, in patients with group E disease (International Classification of Retinoblastoma) or in patients who undergo enucleation and are found to have tumor present at the margin of the optic nerve. If suspected, bone marrow aspiration and lumbar puncture are recommended.[48]
Biopsy
Biopsies are never performed for this disease because of the unacceptable risk of orbital seeding and metastasis.[49] Consequently, diagnosis is based on funduscopic and ultrasonographic characteristics.
Referral
Any child with suspected retinoblastoma should be referred to a specialist ocular oncologist or other ophthalmic specialist. The rarity of the disease, its potential for morbidity and mortality, and the opportunity of a high survival rate with early detection and treatment means that treatment by an inexperienced clinician is inappropriate. Referrals to a pediatric oncologist, radiation oncologist, and geneticist are almost universally handled by the ocular oncologist, who typically has ongoing collaborations with these colleagues.
Use of this content is subject to our disclaimer