Treatment algorithm
Please note that formulations/routes and doses may differ between drug names and brands, drug formularies, or locations. Treatment recommendations are specific to patient groups: see disclaimer
ONGOING
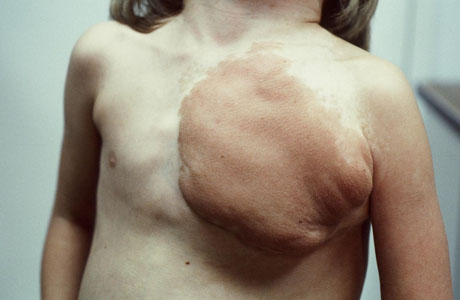
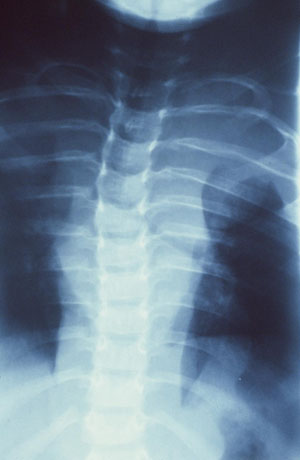
Choose a patient group to see our recommendations
Please note that formulations/routes and doses may differ between drug names and brands, drug formularies, or locations. Treatment recommendations are specific to patient groups. See disclaimer
Use of this content is subject to our disclaimer