Approach
NF1 is a progressive disorder that has a multitude of presentations, varying by age, stage of disease, and variability within the disorder.[12] Clinical and genetic heterogeneity are increasingly a concern in diagnosing apparently mild, regionally localised, or otherwise atypical presentations.
Age
NF1 becomes more obvious and severe with increasing age, largely as a function of increasing numbers and/or size of neurofibromas. A child with minimal manifestation does not exclude an ultimately serious course of the disease. Other serious age-related problems include dystrophic scoliosis and its complications, phaeochromocytoma, and malignant peripheral nerve sheath tumours (MPNSTs).
Stages of disease progress
NF1 is progressive. Its actual manifestations are a function of that progression, best understood in terms of three stages:
Features: the first stage involves those aspects of the disorder most immediately related to the gene mutation itself, such as café au lait spots, neurofibromas, and vertebral dysplasia.
Consequences: the second stage involves those aspects of the disorder deriving from progress of each of the features. For example, dystrophic scoliosis is best understood as a consequence of vertebral dysplasia.
Complications: the third stage involves those aspects of the disorder deriving from progress of each of the consequences. For example, dystrophic scoliosis may lead to spinal cord compression and its attendant neurological compromise.
In terms of treatment options and goals, and future expectations, distinctions between features, consequences, and complications are critical.
Common clinical manifestations
Involvement of the skin, eyes, nervous system, and skeleton should be obvious at presentation. Prompt attention is warranted for pain, neurological deficit, a rapidly enlarging mass, and localised motor or sensory neurological deficit.
Common clinical manifestations include the following:
Skin:
Café au lait spots
Intertriginous freckling
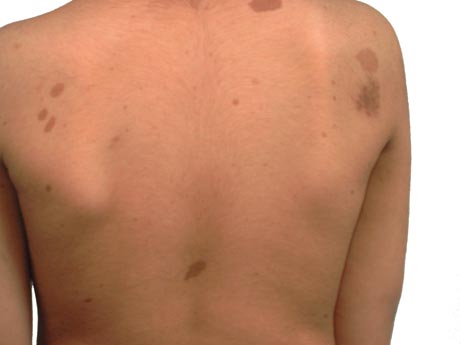
Cutaneous, subcutaneous, and diffuse plexiform neurofibromas [Figure caption and citation for the preceding image starts]: Café au lait spots on the back of a young boyFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Cutaneous neurofibromas in a patient with NF1From the personal collection of Dr D. Gareth Evans; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous neurofibromas in a patient with NF1From the personal collection of Dr D. Gareth Evans; used with permission [Citation ends].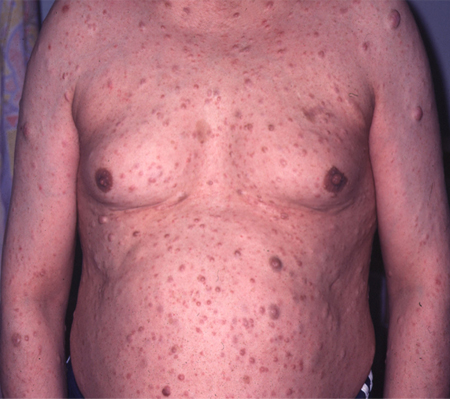 [Figure caption and citation for the preceding image starts]: Left anterior chest diffuse plexiform neurofibroma with overlying hyperpigmentationFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Left anterior chest diffuse plexiform neurofibroma with overlying hyperpigmentationFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].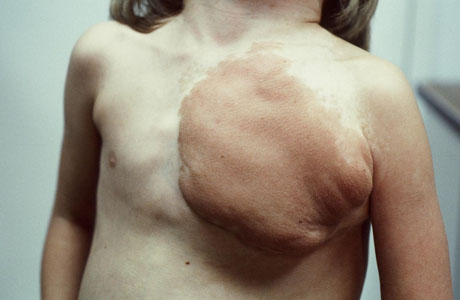 [Figure caption and citation for the preceding image starts]: Radiograph showing extensive involvement of a diffuse plexiform neurofibroma in the upper thorax (bilateral), mediastinum, retroperitoneal space, and omentumFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Radiograph showing extensive involvement of a diffuse plexiform neurofibroma in the upper thorax (bilateral), mediastinum, retroperitoneal space, and omentumFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].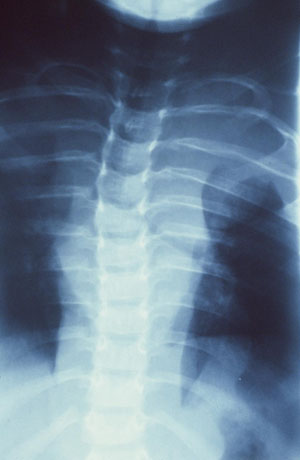
Eyes:
Visual compromise
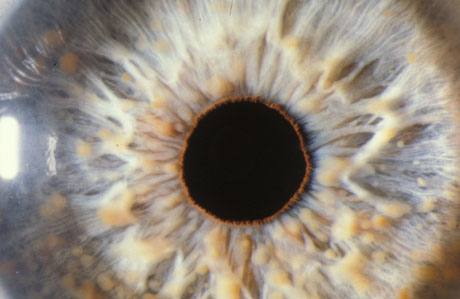
Lisch nodules [Figure caption and citation for the preceding image starts]: Multiple Lisch nodules (pale yellow) on a blue irisFrom the personal collection of Dr Richard A. Lewis; used with permission [Citation ends].
Nervous system:
Learning disability, general incoordination
Seizures
Headache
Skeleton:
Pseudarthrosis of the tibia and/or elsewhere
Scoliosis, which may be dystrophic
Pectus excavatum/carinatum
Genu valgum/varum.
Hypertension may be present if a phaeochromocytoma develops. Most patients with phaeochromocytoma present with continuously or paroxysmally increased blood pressure, or with episodic signs of flushing or palpitations. Although rare, a phaeochromocytoma may also be recognised in an acute setting, in which patients present with an acute abdomen or collapse. About 15% of patients are asymptomatic, and the diagnosis is made coincidentally.[13]
Testing
The diagnosis of NF1 is ordinarily made clinically, although neuroimaging (CT, MRI, PET scan) may be helpful to identify intracranial or paraspinal tumours. Biopsies of café au lait spots are inappropriate unless carried out for mosaic diagnostic testing. Biopsies of neurofibromas for the purpose of confirming the diagnosis of NF1 are only rarely necessary. Biopsies of large neurofibromas to identify an associated MPNST or similar malignancy may be appropriate depending on the presentation.
Molecular testing was previously reserved for problem cases and genetic counselling (e.g., antenatal or pre-implantation diagnosis). However, recommendations from the American Association of Cancer Research recommend RNA-based NF1 mutation testing to confirm the diagnosis. Diagnostic criteria for NF1 and Legius syndrome (NF1-like syndrome) also recommend genetic testing as a criterion to differentiate these from each other and from other disorders (e.g., congenital mismatch repair deficiency) in patients with multiple café au lait patches.[14][15] The increasing evidence for a genotype-phenotype correlation adds further to the utility of testing.
Use of this content is subject to our disclaimer