Approach
There is no treatment for NF1 per se. Treatment depends on the age of the patient, the presence, nature, and severity of manifestations of the disorder, and whether these are features, consequences, or complications. The focus of treatment beyond general support and genetic counselling is limited primarily to surgery. The presence or development of disorders coincidental to NF1 must always be considered.
Phaeochromocytoma
Sustained hypertension - and particularly malignant hypertension - and other indications of a hyperadrenergic state warrant emergent surgical removal of these chromaffin tumours; less severe symptoms also warrant urgent surgery.
Malignant peripheral nerve sheath tumours (MPNSTs)
Require immediate referral to a surgeon and oncologist for extirpation or amputation. These malignancies are generally poorly responsive to chemotherapy or radiotherapy.
Neurofibromas (cutaneous, subcutaneous, diffuse plexiform, nodular plexiform)
These lesions grow slowly, but may distort or impinge on adjacent tissue, and may ultimately cause excessive itching, pain, and/or localised neurological deficits requiring surgery.
Lesions should be observed, with consideration of surgical intervention for selected lesions or targeted systemic therapy. Special attention should be paid to presentation and/or worsening of subcutaneous and nodular plexiform neurofibromas. Concern for the development of a MPNST resulting from both diffuse and nodular is high in the presence of large numbers, or large size, of plexiform neurofibromas.[40]
Specialist care will be required when deciding to proceed to surgery or targeted systemic therapy. Generally, the decision is based on tumour progression and symptoms.
Surgical removal of subcutaneous, non-cutaneous neurofibromas may cause a peripheral neuropathy distal to the surgical disruption of the involved nerve. Palliative surgical removal of nodular plexiform, non-cutaneous neurofibromas may be warranted if very specific goals are delineated beforehand, and where chemotherapy or radiotherapy is not useful.
Targeted systemic therapy with a mitogen-activated protein kinase 1 and 2 (MEK1/2) inhibitor (i.e., selumetinib) may be used for treating symptomatic and inoperable plexiform neurofibromas. The use of MEK inhibitors for large symptomatic plexiform tumours in childhood shows promise.[41] A phase I dose escalation study evaluated 24 children aged 3 to 18 years with inoperable NF1 and plexiform neurofibromas.[42] Treatment with selumetinib demonstrated a dose-dependent partial (≥20% reduction in volume for at least 4 weeks) response in 71% of the patients sustained over a mean of 23 cycles of treatment.[42] No patients experienced disease progression (≥20% increase in volume). One patient discontinued the treatment due to adverse events.[42] The US Food and Drug Administration and European Medicines Agency have granted approval to selumetinib for the treatment of NF1 in children ≥2 years of age (in the US) or ≥3 years of age (in Europe) with symptomatic, inoperable plexiform neurofibromas.[43] In the UK, it is only available under a commercial arrangement agreement.[44]
Café au lait spots and intertriginous freckling neither require nor warrant treatment. Cutaneous, subcutaneous, and diffuse plexiform neurofibromas should be regularly and rigorously observed, with consideration of surgical intervention for selected lesions. Juvenile xanthogranulomas are usually transient and require no treatment.
Pruritus involving the skin is amenable to treatment with mast cell blockers (e.g., ketotifen).[45][46] However, these agents are not commonly used.
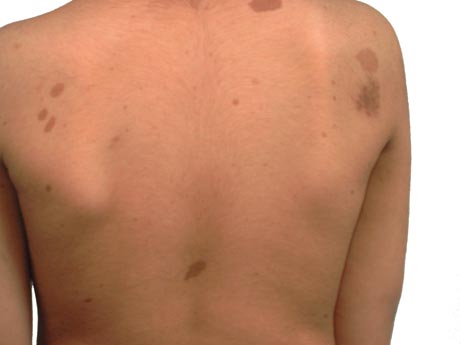
Glomus tumours almost always require surgery once symptomatic.[Figure caption and citation for the preceding image starts]: Café au lait spots on the back of a young boyFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Cutaneous neurofibromas in a patient with NF1From the personal collection of Dr D. Gareth Evans; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous neurofibromas in a patient with NF1From the personal collection of Dr D. Gareth Evans; used with permission [Citation ends].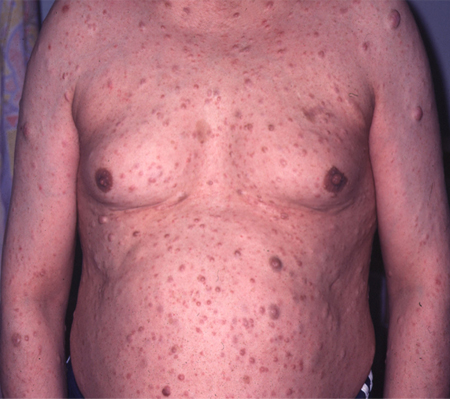 [Figure caption and citation for the preceding image starts]: Left anterior chest diffuse plexiform neurofibroma with overlying hyperpigmentationFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Left anterior chest diffuse plexiform neurofibroma with overlying hyperpigmentationFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].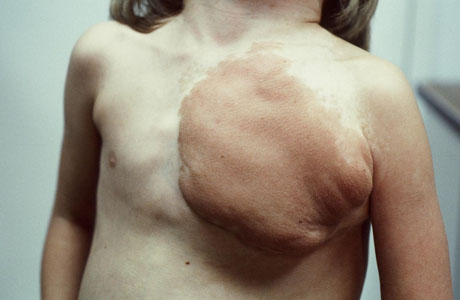 [Figure caption and citation for the preceding image starts]: Radiograph showing extensive involvement of a diffuse plexiform neurofibroma in the upper thorax (bilateral), mediastinum, retroperitoneal space, and omentumFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Radiograph showing extensive involvement of a diffuse plexiform neurofibroma in the upper thorax (bilateral), mediastinum, retroperitoneal space, and omentumFrom the personal collection of Dr Vincent M. Riccardi; used with permission [Citation ends].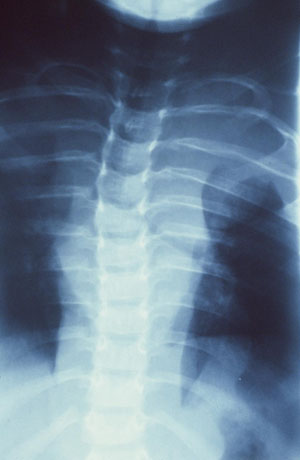
Headache
Headaches are among the most common symptoms of patients with NF1. For the vast majority they are simply coincidental to NF1, but sudden onset of a serious headache warrants consideration of hydrocephalus, intracranial glioma, migraine, or other cerebrovascular compromise. Clinicians must be intimately aware of the patient's headache history.
Nervous system
Learning disability and motor disability should be identified and remediation provided (e.g., through special education resources, learning aids, and possibly central nervous system stimulants).
Therapists, educators, and primary care providers need to communicate regularly regarding progress (both developmental and educational) and update any recommendations.[47]
Patients should be observed closely for indication of an intracranial astrocytoma, including optic glioma and cerebral hemisphere glioma. Optic pathway gliomas causing endocrine/hypothalamic disturbances can be treated with surgery and/or chemotherapy. Surgery is a last resort for cerebral hemisphere glioma.
For progressive gliomas in adults and atypical aggressive low-grade gliomas (LGGs) in children, molecular diagnostics, in addition to NF1, are required for full characterisations of the tumour. ATRX, CDKN2A, and TP53 testing should be conducted. For children, testing of FGFR1 and PIK3CA may also be considered.[48]
Cerebrovascular compromise requires precise anatomical diagnosis and surgical control or ablation as proposed by the consulting neurosurgeon.
Hydrocephalus requires clarification of the pathogenesis (e.g., aqueductal stenosis or external tumour pressure) and treatment appropriate to the pathogenesis, with consideration of shunting.
Treatment of seizures should be appropriate to their nature and frequency according to the recommendations of the consulting neurologist.
Surgical relief of spinal cord compression should be provided if possible and/or accommodation and supportive treatment of the established motor and sensory compromise.
Surgical relief for peripheral neuropathies due to compression from tumour and/or bony collapse should be provided if possible and/or accommodation and supportive treatment of the established motor and sensory compromise. Occasionally, patients with NF1 have peripheral neuropathy that is not secondary to compression.
Eye
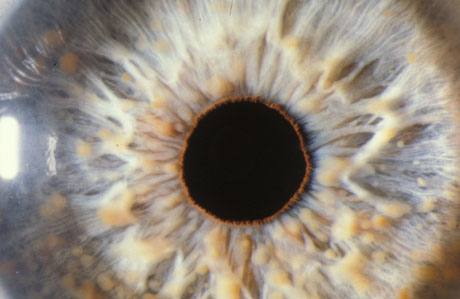
In the first decade and perhaps beyond, patients should be monitored for visual compromise (i.e., due to an optic glioma) and for the telltale diagnostic sign of iris Lisch nodules. Iris Lisch nodules are asymptomatic and do not require treatment.
Unexplained crying and/or irritability in a newborn should raise concern about congenital glaucoma, which is treatable with goniotomy or trabeculotomy. Medical therapy can lower intraocular pressure before and/or after surgery.
Periorbital neurofibromas causing upper or lower lid ptosis will probably require surgery to remove the associated lesion(s).[Figure caption and citation for the preceding image starts]: Multiple Lisch nodules (pale yellow) on a blue irisFrom the personal collection of Dr Richard A. Lewis; used with permission [Citation ends].
Oral
Symptomatic oropharyngeal neurofibromas may require surgery for removal or debulking.
Patients with NF1 appear to be at higher risk for dental caries. Treatment with aggressive follow-up, rigorous hygiene, and restoration should be provided as determined by the dental consultant.
Skeletal
Infants should be carefully assessed for long-bone deformities, especially tibial pseudarthrosis. In the first 5 years, close observation is necessary to identify early vertebral dysplasia and dystrophic scoliosis, as well as sphenoid wing dysplasia. Corrective surgery may be warranted.
Pectus and genu varum/valgum deformities only occasionally require surgical intervention. When the level of severity warrants consideration of treatment, surgery may be the preferred approach, with a timing at the end of the first decade or within the second decade for genu valgum/varum. Orthotics are the primary treatment for genu valgum/varum, ankle valgus, and pes planus.
Vascular
Hypertension must be sought at every clinical visit in all age groups, considering an NF1-related renovascular or phaeochromocytoma origin. Depending on the pathogenesis, both pharmacological treatment and surgery may be warranted as recommended by the paediatric, cardiology, and surgical consultants. Surgical extirpation of the phaeochromocytoma, or stenting, or shunting around the afferent renal vasculature may be warranted.
Gastrointestinal
Observation for constipation/obstipation in children, and bleeding per rectum in older children and adults, is appropriate. When the constipation/obstipation is due to colonic hyperganglionosis, surgery may be warranted. Short of that, closely supervised oral mineral oil may be helpful.
Stromal tumours may cause severe and/or recurrent abdominal pain and/or gastrointestinal bleeding. This should prompt consideration of the need for surgical treatment.
Imatinib, a tyrosine kinase inhibitor, is useful in treating tumours that cannot be surgically removed or that have spread, although the use of imatinib for gastrointestinal stromal tumours (GIST) in NF1 is not as effective as for sporadic GIST.[49][50] It is approved for the treatment of KIT (CD117)-positive GIST.
Sunitinib, a multitargeted tyrosine kinase inhibitor, may be considered if there is disease progression on, or an intolerance to, imatinib.[49][51]
Carcinoid tumours tend to occur in the duodenum and cause facial flushing, diarrhoea, right sided cardiac lesions, facial telangiectasia, and bronchoconstriction.
Haematopoietic
Juvenile chronic myelogenous leukemia is a rare but related manifestation of NF1, and must be considered in any child who develops signs and symptoms suggestive of leukaemia. Aggressive chemotherapy and bone marrow transplantation have been disappointing in improving outcomes.
Psychological
Problems in coping with NF1 and life in general are important aspects of this disorder, especially for children during their school years. Professional counselling and therapy, as well as various forms of self-help group, appear to be beneficial.
Reproductive
Close monitoring of neurofibromas during pregnancy - preferably by an obstetrician familiar with NF1 - is appropriate. All types of neurofibromas typically enlarge during pregnancy, and diffuse plexiform neurofibromas of the pelvis and retroperitoneal space may compromise the later stages of gestation and delivery.
Genetic counselling should be prospective, and antenatal and pre-implantation diagnosis techniques should be made available appropriate to the wishes of the patient and family.
Use of this content is subject to our disclaimer