Aetiology
Graves' disease is an autoimmune condition. The aetiology of thyroid hormone overproduction is stimulation of the thyroid by thyroid-stimulating hormone (TSH) receptor antibodies.[21][22] Although other thyroid antibodies occur in people with Graves' disease (namely, antithyroglobulin [Tg], antithyroid peroxidase [TPO], and antibodies to the sodium iodide transporter), they do not play a part in the development of hyperthyroidism.[23] The fundamental cause of autoimmunity is not clear, but evidence supports a significant genetic component.[3]
Graves' disease is caused by a combination of genetic and environmental factors in an immunologically susceptible individual.[3] The disease concordance rate for monozygotic twins is greater than that reported for dizygotic twins.[24][25] Family and twin studies demonstrate that Graves’ disease is not caused by a single gene defect but has a complex pattern of inheritance. Several gene regions have been linked to Graves' hyperthyroidism, including the human leukocyte antigen (HLA)-DRB1*16:02 allele.[26][27]
Female sex is a risk factor. Moderate alcohol consumption is associated with a reduced risk of Graves' disease, while smoking increases the risk.[28][29][30] Graves’ disease is a common adverse effect of alemtuzumab, a drug used to treat multiple sclerosis.[31]
Pathophysiology
Graves' disease is part of the spectrum of autoimmune thyroid disease that also includes Hashimoto’s thyroiditis (lymphocytic infiltration and destruction of thyroid tissue with secondary antibodies to thyroid peroxidase, thyroglobulin, and other thyroid antigens), painless thyroiditis (subacute destruction of thyroid follicles with release of preformed hormone), and hypothyroidism due to thyroid-stimulating hormone (TSH) receptor-blocking antibodies.[2] Many patients have overlapping autoimmunity; hence the majority of patients with Graves' disease also have elevated antithyroid peroxidase (TPO) antibodies.[2]
Thyroid follicular cells, in response to interferon gamma produced by infiltrating T cells, express HLA class II molecules. This allows presentation of TSH receptors to activated T cells and initiation of the autoimmune cascade. TSH receptor autoantibodies cause thyroid hormone hyperproduction as well as thyroid hypertrophy and hyperplasia of thyroid follicular cells resulting in the clinical manifestations of hyperthyroidism and diffuse goitre.[3]
TSH receptor autoantibody in the serum is measurable by a bioassay utilising cyclic AMP generation in a thyroid cell line (commonly referred to as thyroid-stimulating immunoglobulin [TSI] in the US) or by immunoassays (referred to either as thyrotropin-binding inhibitory immunoglobulin [TBII] or generically as thyrotropin receptor antibody [TRAb]). Activity is correlated with severity of the autoimmune process.[2] Presence of blocking antibodies and co-existing thyroid tissue damage as a component of autoimmune thyroiditis may modify the hyperthyroid state.[3] Some patients have Graves' orbitopathy, dermopathy, and/or acropachy and are euthyroid or even hypothyroid, despite very high levels of TSH receptor antibodies.[10][32]
Extrathyroidal manifestations
The pathogenesis of extrathyroidal manifestations (e.g., orbitopathy, dermopathy, and acropachy) is less clear. TSH receptors are found in a variety of extrathyroidal sites, in particular in retro-orbital and dermal tissue.[3] Fibroblast stimulation by TSH receptor autoantibodies and cytokines produces glycosaminoglycans that trigger orbital fat and tissue expansion.[3] Synergism of insulin-like growth factor 1 (IGF-1) and TSH receptor autoantibodies are implicated in the pathogenesis of orbitopathy (and dermopathy).[3]
The clinical manifestations of orbitopathy (such as proptosis, chemosis, and restrictive ophthalmoplegia) are the consequences of increased orbital soft tissue and extra-ocular muscle volume.[33] Orbital tissues, including muscles, are infiltrated by inflammatory cells, including lymphocytes, mast cells, and macrophages, potentially causing impaired venous return and, in extreme cases, pressure on the optic nerve, and corneal exposure and ulcers.[3][33][34][Figure caption and citation for the preceding image starts]: Lid retraction, mild proptosis, and mild chemosisCourtesy of Dr Vahab Fatourechi [Citation ends].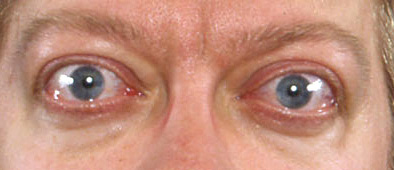 [Figure caption and citation for the preceding image starts]: Orbitopathy and elephantiasisCourtesy of Dr Vahab Fatourechi [Citation ends].
[Figure caption and citation for the preceding image starts]: Orbitopathy and elephantiasisCourtesy of Dr Vahab Fatourechi [Citation ends].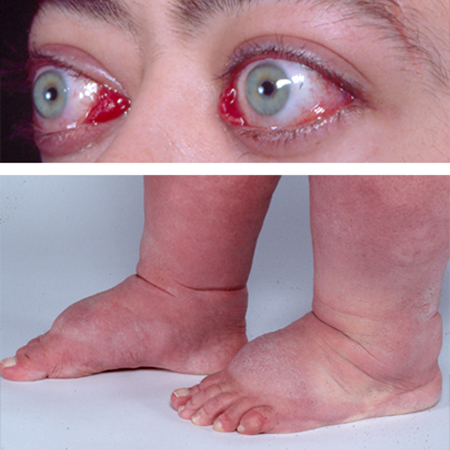
Use of this content is subject to our disclaimer