Common hereditary lysosomal storage diseases
- Overview
- Theory
- Diagnosis
- Management
- Follow up
- Resources
Treatment algorithm
Please note that formulations/routes and doses may differ between drug names and brands, drug formularies, or locations. Treatment recommendations are specific to patient groups: see disclaimer
type 1 Gaucher disease
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
The physician should monitor for skeletal disease, haematological malignancies, endocrine and metabolic abnormalities, Parkinson's disease, and liver cirrhosis, and manage these complications as they arise.
enzyme replacement therapy
Additional treatment recommended for SOME patients in selected patient group
Patients should be treated under consultant supervision. Dose reduction may be possible once patients have been stabilised.
Asymptomatic patients do not warrant treatment.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com However, enzyme replacement therapy (ERT) should be considered in all symptomatic children, and in adults with significant reductions in blood counts (e.g., haemoglobin level <100 g/L [10 g/dL], platelets <100 x 10⁹/L), significant organ enlargement (e.g., spleen size >10x normal), the presence of skeletal disease demonstrated on magnetic resonance imaging, and/or any other organ damage (e.g., evidence of lung damage).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com [100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86. https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com ERT is of demonstrated benefit in improving haematological abnormalities, bone pain, reducing liver and spleen size. Bone density, pulmonary function, and quality of life also improve.[106]Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency - macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991 May 23;324(21):1464-70. http://www.ncbi.nlm.nih.gov/pubmed/2023606?tool=bestpractice.com [107]Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type I Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002 Aug 1;113(2):112-9. http://www.ncbi.nlm.nih.gov/pubmed/12133749?tool=bestpractice.com [108]Gabrowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher disease: phenotypic and genetic variation. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The metabolic and molecular basis of inherited disease. 9th ed. New York, NY: McGraw-Hill; 2006.
Taliglucerase is a plant-derived form of ERT. In 2012, the Food and Drug Administration in the US approved its use for Gaucher disease, but the European Medicines Agency (EMA)'s Committee for Medicinal Products for Human Use (CHMP) recommended against marketing authorisation.
ERT has been associated with serious hypersensitivity reactions, anaphylaxis, and infusion-related reactions. Consider pre-medication with an antihistamine, antipyretic, and/or corticosteroid.
Primary options
imiglucerase: children and adults: 60 units/kg intravenously every 2 weeks initially, adjust according to response
OR
velaglucerase alfa: children ≥4 years of age and adults: 60 units/kg intravenously every 2 weeks initially, adjust according to response
OR
taliglucerase alfa: children ≥4 years of age and adults: 60 units/kg intravenously every 2 weeks initially, adjust according to response
substrate reduction therapy
Additional treatment recommended for SOME patients in selected patient group
This is suitable for less severely affected patients who are unable to tolerate intravenous therapy or who are allergic to available enzyme replacement therapy preparations.
Substrate reduction therapy has been shown to improve anaemia, thrombocytopenia, liver/spleen enlargement, and osteoporosis.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706. http://jama.jamanetwork.com/article.aspx?articleid=2110969 http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com [110]Cox TM, Aerts JM, Andria G, et al. The role of the iminosugar N-butyldeoxynorjirimycin (miglustat) in the management of type 1 (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26(6):513-26. http://www.ncbi.nlm.nih.gov/pubmed/14605497?tool=bestpractice.com [111]Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000 Apr 29;355(9214):1481-5. http://www.ncbi.nlm.nih.gov/pubmed/10801168?tool=bestpractice.com [112]Pastores GM, Giraldo P, Cherin P, et al. Goal-oriented therapy with miglustat in Gaucher disease. Curr Med Res Opin. 2009 Jan;25(1):23-37. http://www.ncbi.nlm.nih.gov/pubmed/19210136?tool=bestpractice.com [113]Poole RM. Eliglustat: first global approval. Drugs. 2014 Oct;74(15):1829-36. http://www.ncbi.nlm.nih.gov/pubmed/25239269?tool=bestpractice.com
Primary options
miglustat: adults: 100 mg orally three times daily, adjust according to response
More miglustatThere are multiple brands of miglustat available; ensure that you use the correct brand for this indication.
OR
eliglustat: adults: dose depends on CYP2D6 genotype and the co-administration of interacting medications; consult specialist for guidance on dose
More eliglustatEstablish patient genotype using a validated test for determining CYP2D6 genotype before starting therapy.[169]National Institute for Health and Care Excellence. Eliglustat for treating type 1 Gaucher disease. Jun 2017 [internet publication]. https://www.nice.org.uk/guidance/hst5
type 2 Gaucher disease
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
Type 2 Gaucher disease is essentially untreatable. The physician should monitor and treat seizures, neurodevelopmental delay, and eye movement disorders.
Enzyme replacement therapy is not effective.
type 3 Gaucher disease
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
The physician should monitor and treat complications of eye movement disorders, neurodevelopmental delay, and skeletal disease.
enzyme replacement therapy
Additional treatment recommended for SOME patients in selected patient group
Asymptomatic patients do not warrant treatment.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com However, enzyme replacement therapy (ERT) should be considered in all symptomatic children, and in adults with significant reductions in blood counts (e.g., haemoglobin level <100 g/L [10 g/dL], platelets <100 x 10⁹/L), significant organ enlargement (e.g., spleen size >10x normal), the presence of skeletal disease demonstrated on magnetic resonance imaging, and/or any other organ damage (e.g., evidence of lung damage).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com [100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86. https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com The visceral and skeletal aspects of the disease respond well to ERT, but neurological manifestations will not be improved as ERT cannot cross the blood-brain barrier.[116]Vellodi A, Tylki-Szymanska A, Davies EH, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009 Oct;32(5):660-4. http://www.ncbi.nlm.nih.gov/pubmed/19655269?tool=bestpractice.com
ERT has been associated with serious hypersensitivity reactions, anaphylaxis, and infusion-related reactions. Consider pre-medication with an antihistamine, antipyretic, and/or corticosteroid.
Primary options
imiglucerase: children and adults: 60 units/kg intravenously every 2 weeks initially, adjust according to response
OR
velaglucerase alfa: children ≥4 years of age and adults: 60 units/kg intravenously every 2 weeks initially, adjust according to response
Fabry disease
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
There are a large number of general supportive aspects in the management of this multisystem disease.
enzyme replacement or chaperone therapy
Additional treatment recommended for SOME patients in selected patient group
Antibody production is provoked by enzyme replacement therapy (ERT) in males, but the impact of this on the efficacy of treatment is unknown. Females do not develop antibodies, perhaps because they are heterozygous and have circulating enzyme.
ERT can slow the progress of organ damage in kidneys and the heart, but these organs may not be returned to normal function. Males should be treated as soon as they are diagnosed; females should be treated if they have symptoms of major organ involvement.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com ERT initiation should be considered in asymptomatic female patients with laboratory, histological, or imaging evidence of kidney, heart, or central nervous system involvement.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
Agalsidase alfa, and agalsidase beta are both effective at preventing renal and cardiovascular complications, compared with no treatment. Agalsidase beta is associated with a lower risk of cerebrovascular complications, compared with agalsidase beta or no treatment.[134]El Dib R, Gomaa H, Ortiz A, et al. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One. 2017 Mar 15;12(3):e0173358. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0173358 http://www.ncbi.nlm.nih.gov/pubmed/28296917?tool=bestpractice.com ERT is effective at reducing pain scores and globotriaosylceramide concentrations in plasma, kidney, and heart.[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
Recommendations have been published on the use of ERT if there is a shortage of this therapy.[170]Linthorst GE, Germain DP, Hollak CE, et al; European Medicines Agency. Expert opinion on temporary treatment recommendations for Fabry disease during the shortage of enzyme replacement therapy (ERT). Mol Genet Metab. 2011 Jan;102(1):99-102. http://www.ncbi.nlm.nih.gov/pubmed/21123099?tool=bestpractice.com These recommendations were published in 2011 when agalsidase beta (one of the two available products for Fabry disease) was undergoing production difficulties.
Pegunigalsidase alfa is another option for treating Fabry disease in adults and has the advantage of a longer plasma half-life.[135]National Institute for Health and Care Excellence. Pegunigalsidase alfa for treating Fabry disease. Oct 2023 [internet publication]. https://www.nice.org.uk/guidance/ta915
ERT has been associated with serious hypersensitivity reactions, anaphylaxis, and infusion-related reactions. Consider pre-medication with an antihistamine, antipyretic, and/or corticosteroid.
Migalastat is an oral chaperone that increases the activity of the endogenous alpha-galactosidase A enzyme in patients with an amenable mutation. Trials have demonstrated the safety and utility of this therapy in patients with amenable mutations, and the treatment is now licensed and available in the US, Europe, Canada, Japan, and several other countries.[137]Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016 Aug 11;375(6):545-55. http://www.nejm.org/doi/full/10.1056/NEJMoa1510198 http://www.ncbi.nlm.nih.gov/pubmed/27509102?tool=bestpractice.com [138]National Institute for Health and Care Excellence. Migalastat for treating Fabry disease. Feb 2017 [internet publication]. https://www.nice.org.uk/guidance/hst4 [139]Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017 Apr;54(4):288-96. [Erratum in: J Med Genet. 2018.] https://jmg.bmj.com/content/54/4/288.long http://www.ncbi.nlm.nih.gov/pubmed/27834756?tool=bestpractice.com [140]Schiffmann R, Bichet DG, Jovanovic A, et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J Rare Dis. 2018 Apr 27;13(1):68. https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0813-7 http://www.ncbi.nlm.nih.gov/pubmed/29703262?tool=bestpractice.com Physicians must confirm that the patient’s mutation is amenable. The manufacturer advises avoidance if the estimated glomerular filtration rate (eGFR) is less than 30 mL/minute/1.73 m². Migalastat is suitable for both naive patients and for patients switching from ERT.[141]Bichet DG, Hopkin RJ, Aguiar P, et al. Consensus recommendations for the treatment and management of patients with Fabry disease on migalastat: a modified Delphi study. Front Med (Lausanne). 2023 Sep 1:10:1220637. https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1220637/full http://www.ncbi.nlm.nih.gov/pubmed/37727761?tool=bestpractice.com
Primary options
agalsidase alfa: children ≥7 years of age and adults: 0.2 mg/kg intravenously every 2 weeks
OR
agalsidase beta: children ≥8 years of age and adults: 1 mg/kg intravenously every 2 weeks
OR
pegunigalsidase alfa: adults: 1 mg/kg intravenously every 2 weeks
OR
migalastat: adults: 123 mg orally once daily on alternate days
TIA and stroke prophylaxis
Treatment recommended for ALL patients in selected patient group
Stroke and transient ischaemic attack (TIA) require careful primary and secondary preventive measures.
pain relief
Treatment recommended for ALL patients in selected patient group
Pain relief with gabapentin or pregabalin is indicated for neuropathic pain. Carbamazepine is also widely used.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com [117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203. http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
Non-steroidal anti-inflammatory drugs should be used sparingly as these patients often have a nephropathy.
Primary options
gabapentin: children: consult specialist for guidance on dose; adults: 1800-3600 mg/day orally given in 3-4 divided doses
OR
pregabalin: children: consult specialist for guidance on dose; adults: 150-300 mg/day orally given in 3 divided doses
OR
carbamazepine: children: consult specialist for guidance on dose; adults: 200-1600 mg/day orally given in 3-4 divided doses
cosmetic surgery or laser treatment
Treatment recommended for ALL patients in selected patient group
Cutaneous lesions may require cosmetic surgery or laser therapy. [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].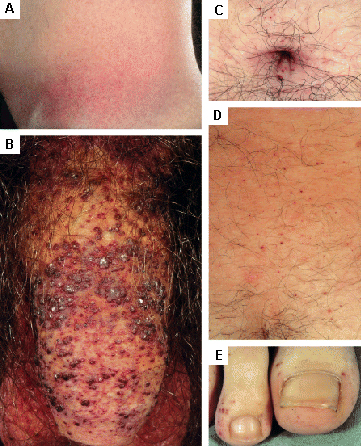 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].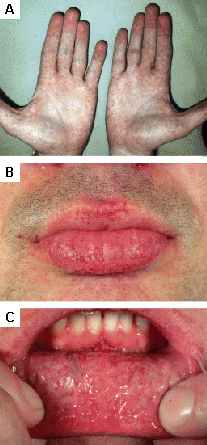
ACE inhibitor or angiotensin-II receptor antagonist
Treatment recommended for ALL patients in selected patient group
Blood pressure and proteinuria should be regularly monitored with early intervention.
Primary options
enalapril: children: consult specialist for guidance on dose; adults: 2.5 to 40 mg orally once daily
OR
irbesartan: children: consult specialist for guidance on dose; adults: 150-300 mg orally once daily
pacemaker + assessment for cardiac surgery
Treatment recommended for ALL patients in selected patient group
Cardiac review is essential in patients with Fabry disease. Early recognition of arrhythmia is important and should be managed with a pacemaker. In addition, surgery, including insertion of pacemakers, septal resection, valve replacement, and even cardiac transplantation should all be considered.[119]Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage diseases. Heart. 2007 Apr;93(4):528-35. http://www.ncbi.nlm.nih.gov/pubmed/17401074?tool=bestpractice.com
mucopolysaccharidosis (MPS)
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
Musculoskeletal problems include spinal deformity, carpal tunnel syndrome, and compression neuropathy. There may be spinal cord compression due to stenosis at the craniocervical region.[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24. http://www.sciencedirect.com/science/article/pii/S1096719212002740 http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com Communicating hydrocephalus and seizures can occur. Patients can also have cardiac valvular disease requiring surgical assessment.
All these disorders require detailed assessment. Physiotherapy, braces, local injections are all possible; surgical therapy may be required.[144]van der Linden MH, Kruyt MC, Sakkers RJ, et al. Orthopaedic management of Hurler's disease after hematopoietic stem cell transplantation: a systematic review. J Inherit Metab Dis. 2011 Jun;34(3):657-69. http://link.springer.com/article/10.1007/s10545-011-9304-x/fulltext.html http://www.ncbi.nlm.nih.gov/pubmed/21416194?tool=bestpractice.com [145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24. http://www.sciencedirect.com/science/article/pii/S1096719212002740 http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
enzyme replacement therapy
Treatment recommended for ALL patients in selected patient group
Enzyme replacement therapy (ERT) is of established benefit in MPS I, II, IVA (Morquio A syndrome), VI, and type VII (Sly syndrome).[148]Hendriksz CJ, Burton B, Fleming TR, et al.; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014 Nov;37(6):979-90. http://link.springer.com/article/10.1007/s10545-014-9715-6/fulltext.html http://www.ncbi.nlm.nih.gov/pubmed/24810369?tool=bestpractice.com [149]Brunelli MJ, Atallah ÁN, da Silva EM. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis type VI. Cochrane Database Syst Rev. 2021 Sep 17;(9):CD009806. https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009806.pub3/full http://www.ncbi.nlm.nih.gov/pubmed/34533215?tool=bestpractice.com [150]Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019 Jun 18;6(6):CD009354. https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009354.pub5/full http://www.ncbi.nlm.nih.gov/pubmed/31211405?tool=bestpractice.com [151]Wikman-Jorgensen PE, López Amorós A, Peris García J, et al. Enzyme replacement therapy for the treatment of Hunter disease: a systematic review with narrative synthesis and meta-analysis. Mol Genet Metab. 2020 Sep - Oct;131(1-2):206-10. http://www.ncbi.nlm.nih.gov/pubmed/32773276?tool=bestpractice.com [152]Gomes DF, Gallo LG, Leite BF, et al. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: systematic review. J Inherit Metab Dis. 2019 Jan;42(1):66-76. http://www.ncbi.nlm.nih.gov/pubmed/30740728?tool=bestpractice.com Various enzymes are approved for these indications. In the UK, the National Institute for Health and Care Excellence recommends elosulfase alfa as an option for treating MPS IVA for people of all ages, and it is only available under a commercial arrangement agreement.[153]National Institute for Health and Care Excellence. Elosulfase alfa for treating mucopolysaccharidosis type 4A: highly specialised technologies guidance. Apr 2022 [internet publication]. https://www.nice.org.uk/guidance/hst19 Phase 3 trials with vestronidase alfa are ongoing.[154]NHS; National Institute for Health Research (NIHR). NIHR Innovation Observatory evidence briefing. Vestronidase alfa (UX-003) for mucopolysaccharidosis type VII (MPS 7; Sly syndrome) NIHRIO (HSRIC) ID: 11463. Apr 2017 [internet publication]. http://www.io.nihr.ac.uk/wp-content/uploads/migrated_new/11463-Vestronidase-alfa-UX-003.pdf [155]ClinicalTrials.gov. A study of UX003 recombinant human beta-glucuronidase (rhGUS) enzyme replacement therapy in subjects with mucopolysaccharidosis type 7, Sly syndrome (MPS 7). Jul 2020 [internet publication]. https://clinicaltrials.gov/ct2/show/NCT02432144
ERT has been associated with serious hypersensitivity reactions, anaphylaxis, and infusion-related reactions. Consider pre-medication with an antihistamine, antipyretic, and/or corticosteroid.
Primary options
MPS I
laronidase: children ≥5 years of age and adults: 0.58 mg/kg intravenously once weekly
OR
MPS II
idursulfase: children ≥5 years of age and adults: 0.5 mg/kg intravenously once weekly
OR
MPS IVA (Morquio A syndrome)
elosulfase alfa: children ≥5 years of age and adults: 2 mg/kg intravenously once weekly
OR
MPS VI
galsulfase: children ≥5 years of age and adults: 1 mg/kg intravenously once weekly
OR
MPS VII
vestronidase alfa: children and adults: 4 mg/kg intravenously every 2 weeks
stem cell transplant
Additional treatment recommended for SOME patients in selected patient group
Stem cell transplantation should be considered for severely affected patients and is of established value in, for example, severe MPS I and MPS VI.[147]Boelens JJ. Trends in hematopoietic stem cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):413-20. http://www.ncbi.nlm.nih.gov/pubmed/16763911?tool=bestpractice.com
Pompe disease
supportive therapy
A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended; care should be coordinated by the centre.
General and supportive care for neonates is multidisciplinary, involving neurologists, anaesthetists, and cardiologists.
Respiratory failure and obstructive sleep apnoea may require different degrees of ventilatory support.
enzyme replacement therapy
Additional treatment recommended for SOME patients in selected patient group
Enzyme replacement therapy (ERT) for children and adults is licensed. Avalglucosidase is an option for late-onset Pompe disease in children 1 year of age and older.
Cipaglucosidase alfa is another option which is approved for the treatment of late-onset Pompe disease in adults who are not improving on their current ERT. Cipaglucosidase alfa is only approved for use in combination with miglustat (an enzyme stabiliser).[165]Blair HA. Cipaglucosidase alfa: first approval. Drugs. 2023 Jun;83(8):739-45. https://link.springer.com/article/10.1007/s40265-023-01886-5 http://www.ncbi.nlm.nih.gov/pubmed/37184753?tool=bestpractice.com
European consensus guidelines recommend an initial 2-year period of ERT for symptomatic adults. ERT may be continued during pregnancy and lactation. Skeletal muscle and respiratory function should be assessed during treatment.[166]van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017 Jun;24(6):768-e31. http://www.ncbi.nlm.nih.gov/pubmed/28477382?tool=bestpractice.com
ERT has been associated with serious hypersensitivity reactions, anaphylaxis, and infusion-related reactions. Consider pre-medication with an antihistamine, antipyretic, and/or corticosteroid.
Primary options
alglucosidase alfa: children and adults: 20 mg/kg intravenously every 2 weeks
OR
avalglucosidase alfa: children ≥1 year of age and <30 kg: 40 mg/kg intravenously every 2 weeks; children ≥1 year of age and ≥30 kg and adults: 20 mg/kg intravenously every 2 weeks
OR
cipaglucosidase alfa: adults ≥40 kg body weight: 20 mg/kg intravenously every 2 weeks
More cipaglucosidase alfaStart 2 weeks after the last enzyme replacement therapy dose. Give 1 hour after miglustat dose (no later than 3 hours after miglustat dose).
and
miglustat: adults ≥40 kg to <50 kg body weight: 195 mg orally every 2 weeks; adults ≥50 kg body weight: 260 mg orally every 2 weeks
More miglustatStart 2 weeks after the last enzyme replacement therapy dose. Give 1 hour before cipaglucosidase alfa dose.
There are multiple brands of miglustat available. Opfolda® is the brand approved for use in Pompe disease, and should be used for this indication.
Tay-Sachs disease
supportive therapy
Palliative care only is required for the infantile form.
Juvenile-onset and chronic or adult-onset forms require supportive care, special needs education, and neurological assessment. Dementia and ataxia are long-term complications that warrant consideration in management.
Enzyme replacement therapy is not available for Tay-Sachs disease.
Niemann-Pick disease
supportive therapy
Palliative care only is required for the severe infantile form of Niemann-Pick type A. Supportive care is indicated for less severe forms.
Niemann-Pick type B typically presents in adults with pulmonary disease and/or hepatosplenomegaly.
Niemann-Pick type C is extremely variable, but neurological assessment is usually important. Enzyme replacement therapy is available for type C.
substrate reduction therapy
Additional treatment recommended for SOME patients in selected patient group
Substrate reduction therapy (SRT) is only available for Niemann-Pick type C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709 http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com SRT with miglustat has been shown to stabilise neurodegenerative symptoms, including dysphagia.[168]Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018 Aug 15;13(1):140. https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0844-0 http://www.ncbi.nlm.nih.gov/pubmed/30111334?tool=bestpractice.com
Primary options
miglustat: adults: 100 mg orally three times daily, adjust according to response
More miglustatThere are multiple brands of miglustat available; ensure that you use the correct brand for this indication.
Choose a patient group to see our recommendations
Please note that formulations/routes and doses may differ between drug names and brands, drug formularies, or locations. Treatment recommendations are specific to patient groups. See disclaimer
Use of this content is subject to our disclaimer