Approach
The lysosomal storage diseases (LSDs) are a highly diverse group of diseases. Initial treatment is general and supportive. A multidisciplinary approach is essential as these are multisystem disorders; many different consultants may be involved in the care of individual patients. Early involvement of specialist centres is recommended, and care should be coordinated by the centre. Specific therapy is not available for many patients (e.g., neonatal type 2 Gaucher disease) and palliation may be the best course.
Enzyme replacement therapy (ERT) has become established for several of the LSDs.[3] In Europe, ERT can be given in the patient's own home for Gaucher, Fabry, Pompe, mucopolysaccharidosis (MPS) I, and MPS II disorders.
Physiotherapists, occupational therapists, clinical nurse consultants, psychologists, teachers, and social workers are all appropriate members of the extended multidisciplinary team.
Gaucher diseases
Type 1 Gaucher disease
Delay in diagnosis continues to be a concern and there is a need to increase awareness among general physicians, paediatricians, and haematologists.[98]
Mildly affected people, for example, normal haemoglobin, platelets >100 x 10⁹/L, minimal enlargement of liver and spleen, no bony lesions on magnetic resonance imaging (MRI), warrant observation but may not need specific therapy. Asymptomatic patients do not warrant treatment.[99]
Splenectomy should be avoided; it will improve blood counts but predisposes to more severe long-term bone disease and to pulmonary hypertension.[100]
There may be skeletal disease leading to bone pain or fracture; orthopaedic surgery may be required to treat the affected joints.
Increased incidence of haematological malignancies in adults should be monitored for: particularly myeloma, but also others such as non-Hodgkin's lymphoma.[100]
Parkinson's disease and liver cirrhosis are other long-term complications requiring monitoring and treatment.
Co-existing endocrine and metabolic abnormalities, including insulin resistance, growth hormone deficiency, and vitamin D insufficiency, should be sought and treated.[101]
ERT with imiglucerase, velaglucerase alfa, and taliglucerase alfa is available.[102][103][104][105]
ERT should be considered in all symptomatic children with Gaucher disease, and in adults with significant reductions in blood counts (e.g., haemoglobin level <100 g/L [10 g/dL], platelets <100 x 10⁹/L), significant organ enlargement (e.g., spleen size >10x normal), the presence of skeletal disease demonstrated on MRI and/or any other organ damage (e.g., evidence of lung damage).[99][100]
ERT has demonstrated benefit in improving haematological abnormalities, bone pain, reducing liver and spleen size. Bone density, pulmonary function, and quality of life also improve.[106][107][108]
Substrate reduction therapy (SRT) has been shown to improve anaemia, thrombocytopenia, liver/spleen enlargement, and osteoporosis.[109] SRT (e.g., miglustat, eliglustat) is an oral therapy that is used in patients who are unable to tolerate ERT or who have difficult venous access.[109][110][111][112][113] Goals of therapy have been recognised for type 1 Gaucher disease and currently available treatments allow most of these to be achieved.[114]
Stem cell transplants have been performed, but ERT is safer.[115]
Type 2 Gaucher disease
Acute neuronopathic type 2 Gaucher disease should only be treated supportively, as premature death is common.
Complications of the disease are seizures, neurodevelopmental delay, and eye movement disorders, which need to be considered as part of the disease management.
Type 2 Gaucher disease is essentially untreatable.
Enzyme replacement therapy is not effective.
Type 3 Gaucher disease
Eye movement disorders, neurodevelopmental delay, and skeletal disease are complications of this disease.
Typically affects children and young adults; the visceral and skeletal aspects of the disease respond well to ERT, but neurological manifestations will not be improved as ERT cannot cross the blood-brain barrier.[116]
Fabry disease
There are a large number of general supportive aspects in the management of this multisystem disease.
Pain relief with gabapentin or pregabalin is indicated for neuropathic pain. Carbamazepine is also widely used.[67][117] Non-steroidal anti-inflammatory drugs should be used sparingly as these patients often have a nephropathy.
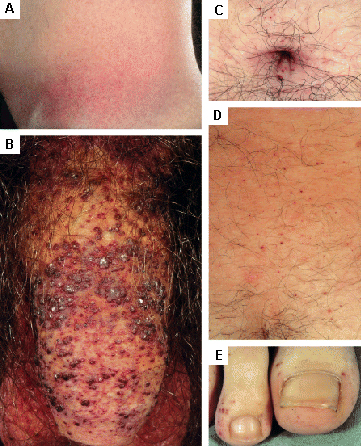
Cutaneous lesions may require cosmetic surgery or laser therapy. [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].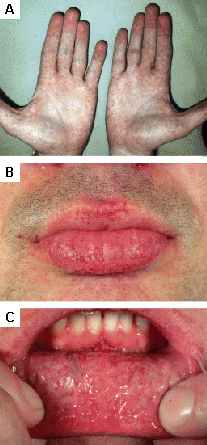
Blood pressure and proteinuria should be regularly monitored with early intervention with ACE inhibitors or angiotensin-II receptor antagonists.[118]
Cardiac review is essential; early recognition of arrhythmia is important. Surgery including insertion of pacemakers, septal resection, valve replacement, and even cardiac transplantation should all be considered.[119]
Stroke and transient ischaemic attack require careful primary and secondary preventive measures.
Gastrointestinal assessment, symptomatic therapy, and endoscopy may all be required to exclude associated conditions.[120]
Depression is common in Fabry disease; the family should be seen together, where possible. Counselling may be necessary.
Deafness is highly likely.[121]
Specific treatment with ERT or chaperone therapy
Guidelines for diagnosis and treatment are available and are regularly updated.[67][117][122][123][124] It is generally agreed that ERT can slow the progress of organ damage in kidneys and the heart, but these organs may not be returned to normal function. Males should be treated as soon as they are diagnosed; females should be treated if they have symptoms of major organ involvement.[67] ERT initiation should be considered in asymptomatic female patients with laboratory, histological, or imaging evidence of kidney, heart, or central nervous system involvement.[67]
Agalsidase alfa and agalsidase beta have demonstrated efficacy and safety in randomised controlled trials.[125][126] Trials and registry data have demonstrated reduced risk of stroke, improvement in cardiac and renal outcomes, improved pain and quality of life, and improved gastrointestinal symptomatology.[127][128][129][130][131][132][133] These agents are safe for use in children and have equal benefit in men and women. Antibody production is provoked by ERT in males, but the impact of this on the efficacy of treatment is unknown. Females do not develop antibodies, perhaps because they are heterozygous and have circulating enzyme. Agalsidase alfa, and agalsidase beta are both effective at preventing renal and cardiovascular complications, compared with no treatment. Agalsidase beta is associated with a lower risk of cerebrovascular complications, compared with agalsidase beta or no treatment.[134] ERT is effective at reducing pain scores and globotriaosylceramide concentrations in plasma, kidney, and heart.[122]
Pegunigalsidase alfa is another option for treating Fabry disease in adults and has the advantage of a longer plasma half-life.[135]
Chaperone therapy consists of small molecules that enhance the activity of the deficient enzyme in patients with residual enzyme activity.[136] These compounds aid intracellular trafficking of the enzyme to improve delivery to the lysosome from the endoplasmic reticulum. It may work in combination with enzyme replacement therapy, and is unaffected by antibodies.
Migalastat is an oral chaperone that increases the activity of the endogenous alpha-galactosidase A enzyme in patients with an amenable mutation. Trials have demonstrated the safety and utility of this therapy in patients with amenable mutations, and the treatment is now licensed and available in the US, Europe, Canada, Japan, and several other countries.[137][138][139][140] Physicians must confirm that the patient’s mutation is amenable. The manufacturer advises avoidance if the estimated glomerular filtration rate (eGFR) is less than 30 mL/minute/1.73 m². Migalastat is suitable for both naive patients and for patients switching from ERT.[141]
Mucopolysaccharidosis (MPS) disorders
General and supportive management
A multidisciplinary approach is essential as MPS is a multisystem disorder. Consultant input should be obtained early.
Early problems can arise with respiration and cardiac disease; special attention must be paid to the airway at all times.[142]
Ear, nose, and throat complications include frequent ear infections.[143]
Musculoskeletal complications are common and input from other specialists, such as orthopaedic surgeons and neurosurgeons, is essential.[144][145] A potentially life-threatening complication, particularly during anaesthesia, is compression of the spinal cord due to stenosis at the craniocervical region.[145] Carpal tunnel syndrome is frequent, as are gibbus deformities.[146]
Careful pre-surgical cardiac assessment should be carried out for all patients given the number of cardiac valvular abnormalities that occur in this group of disorders.
Specific treatment
Stem cell transplantation should be considered for severely affected patients and is of established value in, for example, severe MPS I and MPS VI.[147]
ERT is of established benefit in MPS I, II, IVA (Morquio A syndrome), VI, and type VII (Sly syndrome).[148][149][150][151][152] Various enzymes are approved for these indications. In the UK, the National Institute for Health and Care Excellence recommends elosulfase alfa as an option for treating MPS IVA for people of all ages, and it is only available under a commercial arrangement agreement.[153] Phase 3 trials with vestronidase alfa are ongoing.[154][155][156]
Pompe disease
General and supportive care for neonates is multidisciplinary, involving neurologists, anaesthetists, and cardiologists. ERT for children and adults is licensed.[157]
Systematic reviews have not identified any trials comparing the effectiveness and safety of enzyme replacement therapy for infantile-onset Pompe disease with another intervention, no intervention, or placebo.[158][159] One trial of 18 participants compared two doses of alglucosidase alfa. The trial provided low-quality evidence that long-term alglucosidase alfa treatment markedly extended survival, as well as ventilation-free survival, and improved cardiomyopathy (low-quality evidence); but cardiac function, motor development, and the proportion of children free of invasive ventilation were similar for both doses over a median treatment duration of 2.3 years. There was no significant difference between dose groups.[160] Appropriately powered randomised trials are needed to determine the optimal dose regimen.
ERT for late-onset Pompe disease is associated with significant improvement in walking distance, but not in muscle strength or forced vital capacity.[161] Further prospective trials are needed. Avalglucosidase is an option for late-onset Pompe disease in children 1 year of age and older.[162][163][164] Cipaglucosidase alfa is another option which is approved for the treatment of late-onset Pompe disease in adults who are not improving on their current ERT. Cipaglucosidase alfa is only approved for use in combination with miglustat (an enzyme stabiliser).[165]
European consensus guidelines recommend an initial 2-year period of ERT for symptomatic adults with Pompe disease. ERT may be continued during pregnancy and lactation. Skeletal muscle and respiratory function should be assessed during treatment.[166]
Tay-Sachs disease
Palliative care only is required for the infantile form.
Juvenile-onset and chronic or adult-onset forms require supportive care, special needs education, and neurological assessment. Dementia and ataxia are long-term complications that warrant consideration in management.
ERT is not available for Tay-Sachs disease.
Niemann-Pick diseases
Palliative care only is required for the severe infantile form of Niemann-Pick type A. Supportive care is indicated for less severe forms.
Niemann-Pick type B typically presents in adults with pulmonary disease and/or hepatosplenomegaly. This condition is essentially untreatable. Supportive therapies should be guided by assessments made by a respiratory physician, gastroenterologist, and haematologist.[167]
Niemann-Pick type C is extremely variable, but neurological assessment is usually important. Substrate reduction therapy (SRT) is available for Niemann-Pick type C.[54] SRT with miglustat has been shown to stabilise neurodegenerative symptoms, including dysphagia.[168] Neurological assessment before SRT is important.
Use of this content is subject to our disclaimer