The lysosomal storage diseases (LSDs) are a clinically diverse group of disorders and the differential diagnosis is vast. The different LSDs are easily confused with each other; consultant referral at an early stage is important.
Treatment is now available for many LSDs, and hence there is an increasing emphasis on the need for early diagnosis and intervention before significant organ damage has occurred.
Delay in diagnosis is a significant issue for all LSDs. A diagnosis of an LSD should be considered in cases with relevant clinical features indicative of an LSD.
LSDs are inherited disorders and, therefore, a detailed family history should be taken. The help of a clinical geneticist should be sought if family screening is to be undertaken.
History and examination
Clinical and laboratory diagnosis should be integrated and the possibility of an LSD should be considered in cases with relevant clinical features indicative of an LSD.
Gaucher disease
Type 1 Gaucher disease typically presents in adults with thrombocytopenia and/or splenomegaly.[30]Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017 Feb 17;18(2):441.
https://www.mdpi.com/1422-0067/18/2/441/htm
http://www.ncbi.nlm.nih.gov/pubmed/28218669?tool=bestpractice.com
Fatigue and hepatomegaly are frequent symptoms.[30]Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017 Feb 17;18(2):441.
https://www.mdpi.com/1422-0067/18/2/441/htm
http://www.ncbi.nlm.nih.gov/pubmed/28218669?tool=bestpractice.com
It is more common in Ashkenazi Jews.[10]Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. 2005 Nov-Dec;35(3):355-64.
http://www.ncbi.nlm.nih.gov/pubmed/16185900?tool=bestpractice.com
[11]Beutler E, Nguyen NJ, Henneberger MW, et al. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet. 1993 Jan;52(1):85-8.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1682129/pdf/ajhg00059-0090.pdf
http://www.ncbi.nlm.nih.gov/pubmed/8434610?tool=bestpractice.com
Gaucher disease (severe type 2, acute neuronopathic) presents in neonates with failure to thrive, feeding difficulty, hepatosplenomegaly, abnormal skin, seizures, and gross central nervous system disease; these patients typically die in the first few months of life.[30]Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017 Feb 17;18(2):441.
https://www.mdpi.com/1422-0067/18/2/441/htm
http://www.ncbi.nlm.nih.gov/pubmed/28218669?tool=bestpractice.com
Type 3 (chronic neuronopathic) presents mostly between the ages of 10 and 20 years with an eye movement disorder, hepatosplenomegaly, and bone pain. Seizures, cataracts, cardiac valve disease, joint contractures, and depression may also occur.[41]Grabowski GA. Phenotype, diagnosis, and treatment of Gauchers disease. Lancet. 2008 Oct 4;372(9645):1263-71.
http://www.ncbi.nlm.nih.gov/pubmed/19094956?tool=bestpractice.com
[42]Maas M, Hangartner T, Mariani G, et al. Recommendations for the assessment and monitoring of skeletal manifestations in children with Gaucher disease. Skeletal Radiol. 2008 Mar;37(3):185-8.
http://link.springer.com/article/10.1007/s00256-007-0425-0/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/18094966?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Skeletal magnetic resonance imaging in type 1 Gaucher's disease showing widespread skeletal deposition of substrate with associated necrosis and bony infarction. There is avascular necrosis of the head of the femur (arrow)From the personal collection of Professor Atul B. Mehta [Citation ends].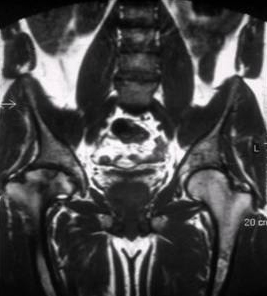
Parkinsonism can occur in association with adult Gaucher disease.[30]Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017 Feb 17;18(2):441.
https://www.mdpi.com/1422-0067/18/2/441/htm
http://www.ncbi.nlm.nih.gov/pubmed/28218669?tool=bestpractice.com
There may be a non-specific history of recurrent respiratory tract infections.
Eye movement disorders, such as delayed initiation of horizontal saccades, are common in infantile disease.[43]Winter AW, Salimi A, Ospina LH, et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol. 2019 Mar;103(3):315-26.
http://www.ncbi.nlm.nih.gov/pubmed/30612093?tool=bestpractice.com
Fabry disease
Fabry disease is often missed in childhood.[38]Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004 Mar;34(3):236-42.
http://www.ncbi.nlm.nih.gov/pubmed/15025684?tool=bestpractice.com
Typical presenting features include burning limb pain, fever, abdominal pain, and diarrhoea.[16]Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003 Nov;162(11):767-72.
http://www.ncbi.nlm.nih.gov/pubmed/14505049?tool=bestpractice.com
[17]Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006 Jan;95(1):86-92.
http://www.ncbi.nlm.nih.gov/pubmed/16498740?tool=bestpractice.com
[44]Zarate AY, Hopkin RJ. Fabry's disease. Lancet. 2008 Oct 18;372(9647):1427-35.
http://www.ncbi.nlm.nih.gov/pubmed/18940466?tool=bestpractice.com
It may present in adulthood with transient ischaemic attack or stroke, cardiac enlargement (hypertrophic cardiomyopathy), and chronic renal failure.[31]Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-30
http://www.ncbi.nlm.nih.gov/pubmed/21092187?tool=bestpractice.com
Corneal clouding, cataracts, hypertension, hypothyroidism, valvular cardiac disease, hearing impairments/sudden deafness, and depression may occur.[12]MacDermot K, Holmes A, Miners A. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001 Nov;38(11):750-60.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1734761/pdf/v038p00750.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11694547?tool=bestpractice.com
Cutaneous lesions are also seen.[31]Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-30
http://www.ncbi.nlm.nih.gov/pubmed/21092187?tool=bestpractice.com
Examples include angiokeratoma and telangiectasia. [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].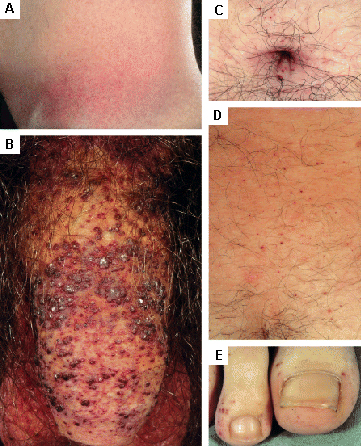 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].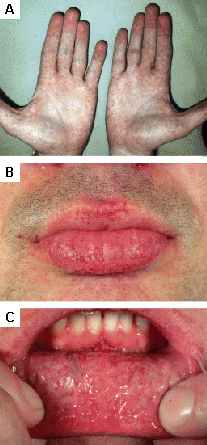
Oral symptoms include dental agenesis, supernumerary teeth, and hyposalivation.[45]Benz K, Hahn P, Hanisch M, et al. Systematic review of oral and craniofacial findings in patients with Fabry disease or Pompe disease. Br J Oral Maxillofac Surg. 2019 Nov;57(9):831-8.
http://www.ncbi.nlm.nih.gov/pubmed/31405600?tool=bestpractice.com
Heterozygous females are frequently (>75% of cases) symptomatic.[36]Wilcox WR, Oliveira JP, Hopkin RJ, et al. Fabry Registrar. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registrar. Mol Genet Metab. 2008 Feb;93(2):112-28.
http://www.ncbi.nlm.nih.gov/pubmed/18037317?tool=bestpractice.com
[37]MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001 Nov;38(11):769-75.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1734754/pdf/v038p00769.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11732485?tool=bestpractice.com
[38]Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004 Mar;34(3):236-42.
http://www.ncbi.nlm.nih.gov/pubmed/15025684?tool=bestpractice.com
[39]Deegan PB, Baehner AF, Barba Romero MA, et al. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006 Apr;43(4):347-52.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2563231
http://www.ncbi.nlm.nih.gov/pubmed/16227523?tool=bestpractice.com
[40]Weidemann F, Niemann M, Sommer C, et al. Interdisciplinary approach towards female patients with Fabry disease. Eur J Clin Invest. 2012 Apr;42(4):455-62.
http://www.ncbi.nlm.nih.gov/pubmed/22049975?tool=bestpractice.com
Mucopolysaccharidosis (severe forms of MPS I, II, and others)
MPS typically presents in the neonatal period or in infancy/early childhood with abnormal facies, large head circumference, hydrocephalus, enlargement of the tongue, hepatosplenomegaly, dysostosis and spinal malformation (including gibbus), corneal clouding, neurodevelopmental delay, joint deformity, and failure to thrive.[5]Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. JAMA. 1999 Jan 20;281(3):249-54.
http://jama.jamanetwork.com/article.aspx?articleid=188380
http://www.ncbi.nlm.nih.gov/pubmed/9918480?tool=bestpractice.com
[46]Muenzer J, Wraith JE, Clarke LA, et al. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009 Jan;123(1):19-29.
http://www.ncbi.nlm.nih.gov/pubmed/19117856?tool=bestpractice.com
Cardiac valve abnormalities, shortness of breath, difficulties in breathing and swallowing also occur.[47]Muenzer J. The mucopolysaccharidoses: a heterogenous group of disorders with variable pediatric presentations. J Pediatr. 2004 May;144(5 suppl):S27-34.
http://www.ncbi.nlm.nih.gov/pubmed/15126981?tool=bestpractice.com
Cognitive impairment is common.
Children with MPS III may present with symptoms of autism spectrum disorder, including language delay and impaired social communication. Symptoms typically emerge at an older age than idiopathic autism spectrum disorder, following a period of normal development.[48]Wolfenden C, Wittkowski A, Hare DJ. Symptoms of autism spectrum disorder (ASD) in individuals with mucopolysaccharide disease type III (Sanfilippo syndrome): a systematic review. J Autism Dev Disord. 2017 Nov;47(11):3620-33.
https://link.springer.com/article/10.1007%2Fs10803-017-3262-6
http://www.ncbi.nlm.nih.gov/pubmed/28856504?tool=bestpractice.com
MPS can also present as recurrent non-immune hydrops fetalis.[1]Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005 Feb;128(4):413-31.
http://www.ncbi.nlm.nih.gov/pubmed/15686451?tool=bestpractice.com
There may be a non-specific history of recurrent respiratory tract infections.[49]Galimberti C, Madeo A, Di Rocco M, et al. Mucopolysaccharidoses: early diagnostic signs in infants and children. Ital J Pediatr. 2018 Nov 16;44(suppl 2):133.
https://ijponline.biomedcentral.com/articles/10.1186/s13052-018-0550-5
http://www.ncbi.nlm.nih.gov/pubmed/30442162?tool=bestpractice.com
Attenuated variants present later (typically age 5 to 15 years), but could present in patients >30 years old with joint deformity, carpal tunnel syndrome, hepatosplenomegaly, compression neuropathy, heart and lung disease, and hearing impairment/sudden deafness.
Pompe disease
Also known as glycogen storage disease type II, the classic infantile form presents in the first few weeks or months of life as feeding difficulties, failure to thrive, hypotonia, and cardiac enlargement with abnormal ECG and echocardiographic changes of hypertrophic cardiomyopathy.[35]van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008 Oct 11;372(9646):1342-53.
http://www.ncbi.nlm.nih.gov/pubmed/18929906?tool=bestpractice.com
[50]Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006 May;148(5):671-6.
http://www.ncbi.nlm.nih.gov/pubmed/16737883?tool=bestpractice.com
[51]Kishnani PS, Steiner RD, Bali D, et al; ACMG Work Group on Management of Pompe Disease. Pompe disease diagnosis and management guideline. Genet Med. 2006 May;8(5):267-88.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3110959
http://www.ncbi.nlm.nih.gov/pubmed/16702877?tool=bestpractice.com
Tongue enlargement, hepatomegaly, and hearing impairment may occur.[45]Benz K, Hahn P, Hanisch M, et al. Systematic review of oral and craniofacial findings in patients with Fabry disease or Pompe disease. Br J Oral Maxillofac Surg. 2019 Nov;57(9):831-8.
http://www.ncbi.nlm.nih.gov/pubmed/31405600?tool=bestpractice.com
Death usually occurs within the first year of life without specific therapy.
Classic feature of Pompe disease is fatigability, found at all ages (e.g., feeding difficulties in infancy, poor sporting performance in childhood, respiratory difficulties, falls, difficulty climbing stairs in adulthood).
There may be a non-specific history of recurrent respiratory tract infections.[52]Hwang B, Meng CC, Lin CY, et al. Clinical analysis of five infants with glycogen storage disease of the heart--Pompe's disease. Jpn Heart J. 1986 Jan;27(1):25-34.
http://www.ncbi.nlm.nih.gov/pubmed/3459901?tool=bestpractice.com
Later-onset attenuated variants present aged 10 to 20 years (may be as late as >30 years) with symptoms of skeletal muscle dysfunction: difficulty climbing stairs, rising from a sitting/lying position, respiratory difficulty, fatigue, joint contracture, and depression.[53]Hagemans ML, Winkel LP, Van Doorn PA, et al. Clinical manifestations and natural course of late-onset Pompe's disease in 54 Dutch patients. Brain. 2005 Mar;128(Pt 3):671-7.
http://brain.oxfordjournals.org/content/128/3/671
http://www.ncbi.nlm.nih.gov/pubmed/15659425?tool=bestpractice.com
Niemann-Pick disease
Presentation will depend on type: hepatosplenomegaly in types A, B, and C; neurodevelopmental delay in type A; and progressive dementia, ataxia or gait disturbance, or hearing problems in type C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709
http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com
[55]Bonnot O, Klünemann HH, Velten C, et al. Systematic review of psychiatric signs in Niemann-Pick disease type C. World J Biol Psychiatry. 2019 Apr;20(4):320-32.
http://www.ncbi.nlm.nih.gov/pubmed/29457916?tool=bestpractice.com
Psychiatric symptoms precede physical symptoms in 75% of patients with type C. Cognitive, memory, and instrumental impairments are the most common psychiatric symptoms, followed by psychosis, altered behaviour, and mood disorders.[55]Bonnot O, Klünemann HH, Velten C, et al. Systematic review of psychiatric signs in Niemann-Pick disease type C. World J Biol Psychiatry. 2019 Apr;20(4):320-32.
http://www.ncbi.nlm.nih.gov/pubmed/29457916?tool=bestpractice.com
Eye movement disorders such as vertical gaze palsies are common in type C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709
http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com
Type A is more common in Ashkenazi Jews.
Tay-Sachs disease
In the infantile form, this presents with hyperacusis and a macular 'cherry red spot'.[56]Chen H, Chan AY, Stone DU, et al. Beyond the cherry-red spot: ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv Ophthalmol. 2014 Jan-Feb;59(1):64-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3864975
http://www.ncbi.nlm.nih.gov/pubmed/24011710?tool=bestpractice.com
The juvenile form presents with optic atrophy (retinitis pigmentosa), progressive dementia and ataxia or gait disturbance, failure to thrive, joint contracture, and depression.
Psychosis, ataxia, dystonia, and cataplexy in young adults may be presenting features of neurodegeneration.[57]Neudorfer O, Pastores GM, Zeng BJ, et al. Late-onset Tay-Sachs disease: phenotypic characterization and genotypic correlations in 21 affected patients. Genet Med. 2005 Feb;7(2):119-23.
https://www.nature.com/articles/gim200521
http://www.ncbi.nlm.nih.gov/pubmed/15714079?tool=bestpractice.com
[58]Rosebush PI, MacQueen GM, Clarke JT, et al. Late-onset Tay-Sachs disease presenting as catatonic schizophrenia: diagnostic and treatment issues. J Clin Psychiatry. 1995 Aug;56(8):347-53.
http://www.ncbi.nlm.nih.gov/pubmed/7635850?tool=bestpractice.com
Eye movement disorders such as vertical gaze palsies are common.
It is more common in Ashkenazi Jews.
Confirmatory diagnosis is by testing the activity of the specific enzyme, or by requesting gene sequence and family studies. It is generally appropriate for generalists to consider the diagnosis and to screen for it; however, early consultant referral is recommended to define the specific diagnosis or to exclude LSDs if there is any doubt.
Enzyme assay
This is the key investigation for most LSDs. It may be appropriate to request assay of a single enzyme (e.g., glucocerebrosidase if Gaucher disease is suspected), or of a group of enzymes (e.g., requesting leukocyte enzyme assay as a screen for MPS disorders). In the X-linked LSDs (e.g., Fabry, MPS II), heterozygous females can often have values that are borderline, as about half of their cells will be normal.
It is also important to request plasma enzyme activity as well as leukocyte activity, as some mutations are associated with disturbed export of enzyme leading to lowered plasma activity with normal leukocyte activity.
Consultant and accredited laboratories should be used as the assays are technically complex and refinements are constantly being made (e.g., to improve diagnosis of Pompe disease).[59]Jack RM, Gordon C, Scott CR, et al. The use of acarbose inhibition in the measurement of acid alpha-glucosidase activity in blood lymphocytes for the diagnosis of Pompe disease. Genet Med. 2006 May;8(5):307-12.
http://www.ncbi.nlm.nih.gov/pubmed/16702881?tool=bestpractice.com
Guidelines for the laboratory diagnosis of MPS VI are now available.[60]Wood T, Bodamer OA, Burin MG, et al. Expert recommendations for the laboratory diagnosis of MPS VI. Mol Genet Metab. 2012 May;106(1):73-82.
http://www.ncbi.nlm.nih.gov/pubmed/22405600?tool=bestpractice.com
These guidelines recommend caution in using urinary glycosaminoglycan (GAG) analysis alone in confirming the diagnosis of MPS VI, and acknowledge enzyme activity analysis as a critical component of diagnosis.
Substrate levels
Increased levels of the appropriate substrate will be detectable in the presence of an enzyme deficiency. Thus, urinary GAGs are elevated in the MPS disorders, and urinary oligosaccharides are elevated in GM1 and GM2 gangliosidosis.[61]Byers S, Rozaklis T, Brumfield LK, et al. Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab. 1998 Dec;65(4):282-90.
http://www.ncbi.nlm.nih.gov/pubmed/9889015?tool=bestpractice.com
Urinary levels of globotriaosylceramide (Gb3) are elevated in Fabry disease. Urinary glucose tetrasaccharide (Glc4) and plasma glucose tetrasaccharide (Hex4) levels are increased in patients with Pompe disease.[62]An Y, Young SP, Kishnani PS, et al. Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Mol Genet Metab. 2005 Aug;85(4):247-54.
http://www.ncbi.nlm.nih.gov/pubmed/15886040?tool=bestpractice.com
Plasma levels of glucosylceramide are elevated in Gaucher disease.
DNA analysis
Genetic studies can provide confirmation of the diagnosis for most LSDs.
In Gaucher disease, there are 6 common mutations seen among Ashkenazi Jews, and a small number of other recurrent mutations occur such that diagnostic kits are available allowing detection of common mutations by polymerase chain reaction (PCR).[63]Beutler E, Gelbart T, Kuhl W, et al. Mutations in Jewish patients with Gaucher disease. Blood. 1992 Apr 1;79(7):1662-6.
https://www.sciencedirect.com/science/article/pii/S0006497120736578
http://www.ncbi.nlm.nih.gov/pubmed/1558964?tool=bestpractice.com
[64]Velayati A, Knight MA, Stubblefield BK, et al. Identification of recombinant alleles using quantitative real-time PCR implications for Gaucher disease. J Mol Diagn. 2011 Jul;13(4):401-5.
https://www.jmdjournal.org/article/S1525-1578(11)00093-6/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/21704274?tool=bestpractice.com
Often only one mutant allele is detected; therefore, clinical manifestations should be considered in differentiating carriers from sufferers.
In Fabry disease, most classically affected males have 'private' mutations (rare mutations typically found only in small populations) that are typically null (associated with absent enzyme).[31]Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-30
http://www.ncbi.nlm.nih.gov/pubmed/21092187?tool=bestpractice.com
In Pompe disease, the MPS disorders, and most other LSDs, a large number of mutations are reported for each disease, but there are recurrent mutations that are typically more common in some communities than in others.
The DNA result needs to be considered in light of detailed clinical information and data on enzyme activity.
Some mutations are polymorphisms and may not be associated with disease. Many PCR tests only screen for common mutations; 'private' mutations are only detected by sequence analysis in research laboratories.
Denaturing high-pressure liquid chromatography is a method for rapid screening for single-base mutations.
Carrier detection in X-linked disorders can only be reliably achieved by DNA analysis.
Not all mutations have been analysed and some disorders will only be diagnosed in consultant laboratories; it is essential that specialist centres are consulted in the design and implementation of the diagnostic strategy for these rare diseases.
Full blood count
Anaemia may be multifactorial due to chronic disease, renal impairment, feeding difficulties, or marrow replacement.
Leukopenia is typically due to splenomegaly. Abnormal inclusions may be seen in leukocytes: for example, periodic acid-Schiff-positive lymphocytes in Pompe disease; abnormal leukocyte appearances in blood/marrow in Tay-Sachs.[65]Noonan SM, Weiss L, Riddle JM. Ultrastructural observations of cytoplasmic inclusions in Tay-Sachs lymphocytes. Arch Pathol Lab Med. 1976 Nov;100(11):595-600.
http://www.ncbi.nlm.nih.gov/pubmed/185985?tool=bestpractice.com
Thrombocytopenia is typically due to splenomegaly, although there may be disturbed platelet function in, for example, Hermansky-Pudlak syndrome.[66]Salles II, Feys HB, Iserbyt BF, et al. Inherited traits affecting platelet function. Blood Rev. 2008 May;22(3):155-72.
http://www.ncbi.nlm.nih.gov/pubmed/18180086?tool=bestpractice.com
ECG and echocardiogram
These are essential for cardiac assessment in infantile Pompe, adult Fabry, and many neonatal MPS disorders.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27.
https://www.sciencedirect.com/science/article/pii/S1096719217307680
http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
[68]Nair V, Belanger EC, Veinot JP. Lysosomal storage disorders affecting the heart: a review. Cardiovasc Pathol. 2019 Mar - Apr;39:12-24.
http://www.ncbi.nlm.nih.gov/pubmed/30594732?tool=bestpractice.com
Findings may indicate enlarged cardiac chambers, abnormal valves, and functional defects in these patients.
Pulmonary function tests
These are critical in Niemann-Pick types A and B for assessing severity. Abnormal lung function with deficient gas transfer will be detected in these patients.[69]von Ranke FM, Pereira Freitas HM, Mançano AD, et al. Pulmonary involvement in Niemann-Pick disease: a state-of-the-art review. Lung. 2016 Aug;194(4):511-8.
http://www.ncbi.nlm.nih.gov/pubmed/27164983?tool=bestpractice.com
Ophthalmic examination
Macular 'cherry spot' and optic atrophy or retinitis pigmentosa are seen in Tay-Sachs.[56]Chen H, Chan AY, Stone DU, et al. Beyond the cherry-red spot: ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv Ophthalmol. 2014 Jan-Feb;59(1):64-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3864975
http://www.ncbi.nlm.nih.gov/pubmed/24011710?tool=bestpractice.com
Corneal clouding is seen in MPS disorders, Fabry disease, and some Gaucher disease.[43]Winter AW, Salimi A, Ospina LH, et al. Ophthalmic manifestations of Gaucher disease: the most common lysosomal storage disorder. Br J Ophthalmol. 2019 Mar;103(3):315-26.
http://www.ncbi.nlm.nih.gov/pubmed/30612093?tool=bestpractice.com
Characteristic cataract seen in Fabry disease with tortuous retinal vessels.[56]Chen H, Chan AY, Stone DU, et al. Beyond the cherry-red spot: ocular manifestations of sphingolipid-mediated neurodegenerative and inflammatory disorders. Surv Ophthalmol. 2014 Jan-Feb;59(1):64-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3864975
http://www.ncbi.nlm.nih.gov/pubmed/24011710?tool=bestpractice.com
Cataracts are also seen in Gaucher disease, MPS disorders, and other LSDs.
Biopsy of affected tissue
This should only be considered after less invasive tests have been undertaken. Tissue biopsy of an organ that is enlarged and/or has disordered function (e.g., liver, kidney, heart) can be done to demonstrate increased levels of substrate, often associated with cellular engorgement.
Bone marrow biopsy is often undertaken in the investigation of hepatosplenomegaly and may reveal characteristic storage cells such as substrate-laden macrophages in Gaucher disease.[70]Pandey MK, Grabowski GA. Immunological cells and functions in Gaucher disease. Crit Rev Oncog. 2013;18(3):197-220.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3661296
http://www.ncbi.nlm.nih.gov/pubmed/23510064?tool=bestpractice.com
Electron microscopy of affected tissue may show characteristic changes of lysosomal damage. Muscle biopsy is of diagnostic utility in many LSDs (e.g., late-onset Pompe; however, appearances may be normal); skin biopsy is often favoured as it is relatively non-invasive and cultured skin fibroblasts are useful for enzyme assay.[71]Vissing J, Lukacs Z, Straub V. Diagnosis of Pompe disease: muscle biopsy vs blood-based assays. JAMA Neurol. 2013 Jul;70(7):923-7.
http://www.ncbi.nlm.nih.gov/pubmed/23649721?tool=bestpractice.com
[72]Alroy J, Ucci AA. Skin biopsy: a useful tool in the diagnosis of lysosomal storage diseases. Ultrastruct Pathol. 2006 Nov-Dec;30(6):489-503.
http://www.ncbi.nlm.nih.gov/pubmed/17182441?tool=bestpractice.com
[73]Lakomá J, Donadio V, Liguori R, et al. Characterization of human dermal fibroblasts in Fabry disease. J Cell Physiol. 2016 Jan;231(1):192-203.
http://www.ncbi.nlm.nih.gov/pubmed/26058984?tool=bestpractice.com
The decision of the appropriate biopsy tissue will be undertaken by the consultant.[Figure caption and citation for the preceding image starts]: Bone marrow aspirate showing a typical Gaucher's cell. This is a macrophage that has ingested cellular material; the undegraded substrate (glucosyl ceramide) accumulates within lysosomesFrom the personal collection of Professor Atul B. Mehta [Citation ends].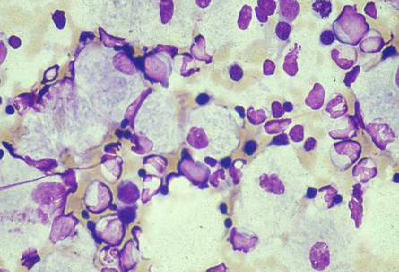 [Figure caption and citation for the preceding image starts]: Electron microscope image of biopsy of pulmonary epithelial cells in Fabry's disease showing the characteristic deposits of substrate in lysosomes, forming 'zebra bodies': (A) magnification x8000, (B) magnification x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Electron microscope image of biopsy of pulmonary epithelial cells in Fabry's disease showing the characteristic deposits of substrate in lysosomes, forming 'zebra bodies': (A) magnification x8000, (B) magnification x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; used with permission [Citation ends].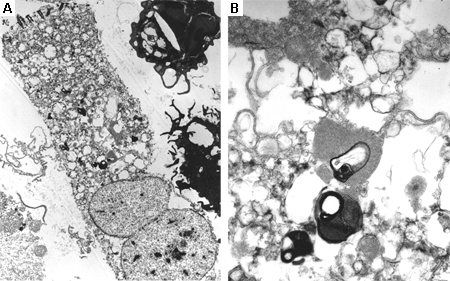
Imaging
LSDs are diverse multisystem disorders and many different forms of imaging are important and relevant. Examples are computed tomography (CT) and magnetic resonance imaging (MRI) to measure organ volumes and to assess the skeleton in Gaucher disease; echocardiogram, ultrasound, or MRI to assess heart, kidneys, and brain in Fabry disease; and CT and x-ray to assess communicating hydrocephalus, atlanto-axial dysplasia, and spinal/vertebral anomalies in MPS disorders.[42]Maas M, Hangartner T, Mariani G, et al. Recommendations for the assessment and monitoring of skeletal manifestations in children with Gaucher disease. Skeletal Radiol. 2008 Mar;37(3):185-8.
http://link.springer.com/article/10.1007/s00256-007-0425-0/fulltext.html
http://www.ncbi.nlm.nih.gov/pubmed/18094966?tool=bestpractice.com
[74]Simpson WL, Hermann G, Balwani M. Imaging of Gaucher disease. World J Radiol. 2014 Sep 28;6(9):657-68.
https://www.wjgnet.com/1949-8470/full/v6/i9/657.htm
http://www.ncbi.nlm.nih.gov/pubmed/25276309?tool=bestpractice.com
[75]Perry R, Shah R, Saiedi M, et al. The role of cardiac imaging in the diagnosis and management of Anderson-Fabry Disease. JACC Cardiovasc Imaging. 2019 Jul;12(7 pt 1):1230-42.
https://www.sciencedirect.com/science/article/pii/S1936878X19304619
http://www.ncbi.nlm.nih.gov/pubmed/31272606?tool=bestpractice.com
[76]Ginsberg L, Manara R, Valentine AR, et al. Magnetic resonance imaging changes in Fabry disease. Acta Paediatr Suppl. 2006 Apr;95(451):57-62.
http://www.ncbi.nlm.nih.gov/pubmed/16720467?tool=bestpractice.com
[77]Spina V, Barbuti D, Gaeta A, et al. The role of imaging in the skeletal involvement of mucopolysaccharidoses. Ital J Pediatr. 2018 Nov 16;44(suppl 2):118.
https://ijponline.biomedcentral.com/articles/10.1186/s13052-018-0556-z
http://www.ncbi.nlm.nih.gov/pubmed/30442151?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Skeletal magnetic resonance imaging in type 1 Gaucher's disease showing widespread skeletal deposition of substrate with associated necrosis and bony infarction. There is avascular necrosis of the head of the femur (arrow)From the personal collection of Professor Atul B. Mehta [Citation ends].
Significance of negative test results
Early consultant referral is necessary. The clinician should liaise closely with the laboratory and ensure that samples reach the laboratory quickly, to avoid decay and possible false negative interpretation. Some assays are being refined (e.g., for Pompe disease); some DNA/enzyme tests may only be available through consultant referral to international research laboratories.[78]Zuckerman S, Lahad A, Shmueli A, et al. Carrier screening for Gaucher disease: lessons for low-penetrance, treatable diseases. JAMA. 2007 Sep 19;298(11):1281-90.
http://jamanetwork.com/journals/jama/fullarticle/208859?resultclick=1
http://www.ncbi.nlm.nih.gov/pubmed/17878420?tool=bestpractice.com
Deficiency of a sphingolipid activator protein or saposin may result in a clinical phenotype that resembles an LSD and may underlie Gaucher disease and metachromatic leukodystrophy.[1]Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005 Feb;128(4):413-31.
http://www.ncbi.nlm.nih.gov/pubmed/15686451?tool=bestpractice.com