Aetiology
Lysosomal storage diseases (LSDs) are single gene disorders usually due to inherited defects in synthesis (rarely defective activation, targeting, or intra-cellular trafficking) of lysosomal enzymes.[2]
These enzymes are crucial for acidic hydrolysis of extracellular macromolecules (e.g., glycosphingolipids derived from breakdown of membranes of blood cells, gangliosides derived from neurons, and glycosaminoglycans derived from connective tissue and extracellular matrix). Substantial substrate also derives from intracellular turnover of macromolecules.
Most LSDs are autosomal recessive; however, 3 of the diseases (Fabry, mucopolysaccharidosis [MPS] II, and Danon's) are sex-linked. There is considerable phenotypic heterogeneity among all LSDs, and genotype-phenotype correlations are generally poor. However, in general, the lower the residual enzyme activity, the greater the accumulation of substrate, the younger the age of onset, and the more severe the disease.[1][21]
Gene deletions, nonsense mutations, and mutations that severely disrupt the tertiary structure of the enzyme are generally incompatible with life (e.g., as observed in type 2 Gaucher disease) or associated with severe manifestations. Mutations associated with residual enzyme activity have a high level of phenotypic heterogeneity, and even individual siblings with the same mutation can have widely divergent clinical manifestations.
Neurological involvement in Gaucher disease is not seen with patients who have at least one allele with the N370S mutation (common in Ashkenazi Jews), as this is sufficient to give a residual enzyme activity that 'protects' the central nervous system (CNS).[22][23]
Sphingolipidoses
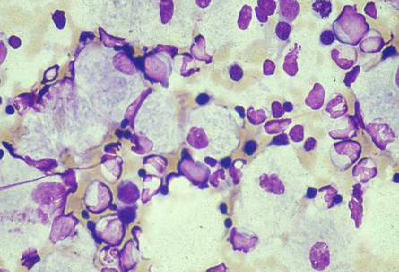
Gaucher disease (beta-glucocerebrosidase deficiency) type 1 (most common form), types 2 and 3 (with CNS involvement) [Figure caption and citation for the preceding image starts]: Bone marrow aspirate showing a typical Gaucher's cell. This is a macrophage that has ingested cellular material; the undegraded substrate (glucosyl ceramide) accumulates within lysosomesFrom the personal collection of Professor Atul B. Mehta [Citation ends].
 [Figure caption and citation for the preceding image starts]: Skeletal magnetic resonance imaging in type 1 Gaucher's disease showing widespread skeletal deposition of substrate with associated necrosis and bony infarction. There is avascular necrosis of the head of the femur (arrow)From the personal collection of Professor Atul B. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Skeletal magnetic resonance imaging in type 1 Gaucher's disease showing widespread skeletal deposition of substrate with associated necrosis and bony infarction. There is avascular necrosis of the head of the femur (arrow)From the personal collection of Professor Atul B. Mehta [Citation ends].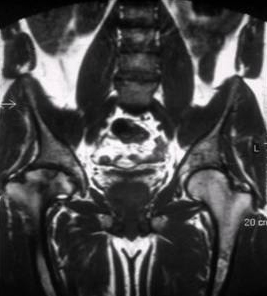
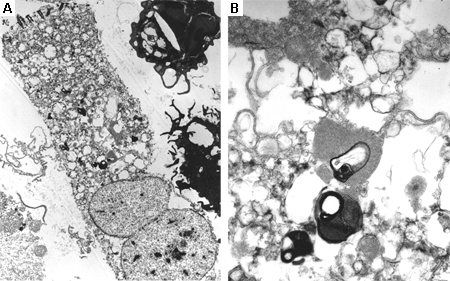
Fabry disease (alpha-galactosidase deficiency)[Figure caption and citation for the preceding image starts]: Electron microscope image of biopsy of pulmonary epithelial cells in Fabry's disease showing the characteristic deposits of substrate in lysosomes, forming 'zebra bodies': (A) magnification x8000, (B) magnification x62,500Kelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; used with permission [Citation ends].
 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].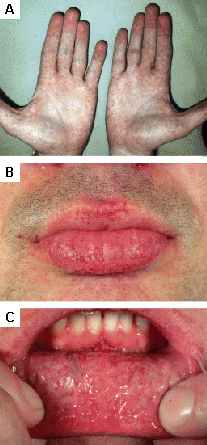 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].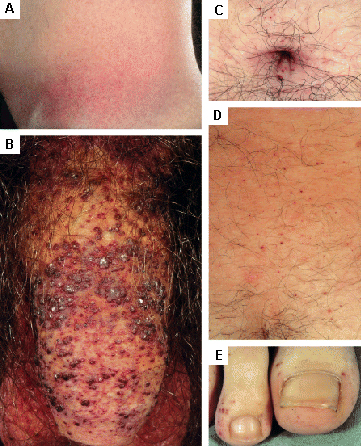
Tay-Sachs (beta-hexosaminidase A deficiency)
Sandhoff's (beta-hexosaminidase B deficiency)
Krabbe's (beta-galactosylceramidase deficiency)
Niemann-Pick types A, B (acid sphingomyelinase deficiency)
Niemann-Pick type C (complex transport defect)
Metachromatic leukodystrophy (arylsulphatase A deficiency)
GM1 gangliosidosis (beta-galactosidase deficiency)
Mucopolysaccharidosis (MPS)
MPS I (alpha-L-iduronidase deficiency) severe form (Hurler's), milder forms (Hurler-Scheie, Scheie's)
MPS II/Hunter's syndrome (iduronate-2-sulphatase deficiency).
MPS III/Sanfilippo's syndrome (4 subtypes: A, B, C, D) (deficiency of heparan-N-sulphatase and other enzymes)
MPS IV/Morquio's syndrome (2 subtypes: A, B) (N-acetylgalactosamine 6-sulphatase and beta-galactosidase deficiency)
MPS VI/Maroteaux-Lamy syndrome (N-acetylgalactosamine 4-sulphatase deficiency)
MPS VII/Sly's syndrome (beta-glucuronidase deficiency)
MPS IX/Natowicz syndrome (hyaluronidase deficiency)
MPS X/Arylsulfatase K deficiency
Other
Pompe disease/glycogen storage disease type 2 (acid maltase deficiency).
Glycoproteinoses (e.g., aspartylglucosaminuria, fucosidosis, alpha- and beta-mannosidosis, Schindler's disease)
Multiple sulphatase deficiency, mucolipidosis II/III, IV, Danon's disease, Wolman's disease, lysosomal transport defects (e.g., cystinosis, sialic acid storage disease)
Pathophysiology
Enzyme deficiency leads to disturbed intermediary metabolism with accumulation of undegraded substrate.[1][2] This not only leads to enlargement of individual cells and organs but also causes secondary biochemical and structural abnormalities. Lysosomal hypertrophy occurs, leading to gross disturbance of lysosomal function.
Neuronal changes are partly structural and due to increased storage; but accelerated apoptosis of neurons has been demonstrated in the mucopolysaccharidosis disorders, and disturbed calcium metabolism resulting in excessive neuronal cell death has been demonstrated in neuronopathic Gaucher disease.[24][25]
Extralysosomal accumulation of substrate can disrupt membrane function, mitochondrial metabolism, membrane ion channels and transporters, and signal transduction across membrane 'lipid rafts'.[1][26]
Macrophage activation, possibly mediated by cytokine release, has been demonstrated in Gaucher disease and in other lysosomal storage diseases (LSDs).[27][28] Accumulation of lysosphingolipids (i.e., glycosphingolipids that do not have N-acylated fatty acids) may be of pathogenical significance in Krabbe's, Gaucher, and Fabry diseases.[29]
Disease-specific pathophysiology
Gaucher disease: due to beta-glucocerebrosidase deficiency. This renders the lysosome unable to break down glycosphingolipids, leading to an accumulation of glucosylceramides.[30]
Fabry disease: caused by alpha-galactosidase deficiency, which results in accumulation of globotriaosylceramide within lysosomes.[31]
Tay-Sachs disease: results from beta-hexosaminidase A deficiency. Gangliosides accumulate in lysosomes, particularly in neurons.[32]
Niemann-Pick disease (NPD): types A and B are due to acid sphingomyelinase deficiency. NPD type A is a severe neurodegenerative disorder that typically presents in infancy, while NPD type B, which usually spares the nervous system, is characterised by substrate accumulation in the reticuloendothelial system (causing hepatosplenomegaly, pulmonary disease) and presents later. NPD type C is a complex disorder of intracellular transport and processing of cholesterol, which usually presents in childhood with ataxia, disordered eye movements, and learning difficulties, but can present in adults as a neurological illness or with hepatosplenomegaly.[33]
Mucopolysaccharidoses: characterised by incomplete breakdown, and progressive accumulation, of glycosaminoglycans.[34]
Pompe disease: caused by acid maltase deficiency, leading to accumulation of glycogen in lysosomes.[35]
Classification
Sphingolipidoses
Incidence: >1/50,000 people[5]
Gaucher disease (beta-glucocerebrosidase deficiency) type 1 (most common form), types 2 and 3 (with central nervous system involvement)
Fabry disease (alpha-galactosidase deficiency)
Incidence: <1/100,000 people[5]
Tay-Sachs (beta-hexosaminidase A deficiency)
Sandhoff's (beta-hexosaminidase B deficiency)
Krabbe's (beta-galactosylceramidase deficiency)
Niemann-Pick types A, B (acid sphingomyelinase deficiency)
Niemann-Pick type C (complex transport defect)
Metachromatic leukodystrophy (arylsulphatase A deficiency)
GM1 gangliosidosis (beta-galactosidase deficiency)
Mucopolysaccharidosis (MPS)
Incidence: >1/100,000 people[5]
MPS I (alpha-L-iduronidase deficiency) severe form (Hurler's), milder forms (Hurler-Scheie, Scheie's)
MPS II/Hunter syndrome (iduronate-2-sulphatase deficiency)
Incidence: <1/100,000 people[5]
MPS III/Sanfilippo's syndrome (4 subtypes: A, B, C, D) (deficiency of heparan-N-sulphatase and other enzymes)
MPS IV/Morquio's syndrome (2 subtypes: A, B) (N-acetylgalactosamine 6-sulphatase and beta-galactosidase deficiency)
MPS VI/Maroteaux-Lamy syndrome (N-acetylgalactosamine 4-sulphatase deficiency)
MPS VII/Sly's syndrome (beta-glucuronidase deficiency)
MPS IX/Natowicz syndrome (hyaluronidase deficiency)
MPS X/Arylsulfatase K deficiency
Other
Incidence: >1/50,000 people[5]
Pompe disease/glycogen storage disease type 2 (acid maltase deficiency)
Incidence: >1/100,000 people[5]
Glycoproteinoses (e.g., aspartylglucosaminuria, fucosidosis, alpha- and beta-mannosidosis, Schindler's disease)
Multiple sulphatase deficiency, mucolipidosis II/III, IV, Danon's disease, Wolman's disease, lysosomal transport defects (e.g., cystinosis, sialic acid storage disease)
Use of this content is subject to our disclaimer