History and exam
Key diagnostic factors
common
presence of risk factors
Key risk factors include Ashkenazi ethnicity for many lysosomal storage diseases (LSDs) and male sex for some LSDs (mucopolysaccharidosis II, Fabry diseases). Some present in childhood and others in adulthood. Type 1 (adult-onset) Gaucher disease, Niemann-Pick type A disease, and Tay-Sachs disease are more common among Ashkenazi Jews.
Family history
These are inherited disorders and should be assessed after appropriate consultation with a clinical geneticist. Occasionally patients do not have a family history, either because the families are small (these are mainly autosomal-recessive disorders and carriers are generally asymptomatic), or because it may be a new mutation. Family history in the extended pedigree is common in X-linked lysosomal storage diseases (Fabry, mucopolysaccharidosis [MPS] II, and Danon's).
onset in childhood (MPS, Pompe, Gaucher, Fabry, Niemann-Pick type A)
Age <1 year: the severe forms of the mucopolysaccharidosis (MPS) disorders, classic Pompe disease, and neuronopathic forms of Gaucher disease present in early infancy.[30][35][46][50][79] Early age of onset is a strong indicator of a severe long-term course, and is often associated with low residual enzyme activity.
Age 1 to 10 years: most MPS disorders (including attenuated variants) and Niemann-Pick type A will present by this age.[80] Most males with classic Fabry disease will have had some symptoms, although the average age of diagnosis in males is older.[38]
onset in adolescence (Fabry, Pompe, Gaucher types 1, 3, mucopolysaccharidosis, Niemann-Pick types B, C)
Age 10 to 20 years: most type 3 Gaucher disease; most of the more severe type 1 Gaucher disease but not all; most classic Fabry disease; and most Pompe, including adult onset, will be symptomatic, although not all diagnosed at these ages. Niemann-Pick types B and C are variable; severe phenotypes present earlier, milder phenotypes present later. Attenuated forms of mucopolysaccharidosis disorders are also highly variable.
onset in adulthood (Fabry, Gaucher type 1, Pompe)
Age 20 to 50 years: typical age of onset for most patients with type 1 Gaucher disease, and the age that patients with classic Fabry disease develop clinical problems.
Age >50 years: lysosomal storage diseases are unlikely to present in older age; exceptions are milder forms of type 1 Gaucher disease, atypical Fabry disease, and some patients with milder Pompe disease.
hepatomegaly and/or splenomegaly
Hepatomegaly is seen in type 2 and 3 Gaucher, mucopolysaccharidosis disorders, Pompe, and Niemann-Pick diseases.[5][30][46][49][54]
Splenomegaly is seen in type 2 and 3 Gaucher, mucopolysaccharidosis disorders, and Niemann-Pick diseases.[5][30][46][49][54]
Hepatosplenomegaly is common in mucopolysaccharidosis disorders, Gaucher disease, and Niemann-Pick types A, B, and C.[5][30][46][49][54]
hyperacusis
Characteristic of Tay-Sachs disease.[81]
history of renal failure
Found in adult Fabry disease.[31]
skin rash/cutaneous lesions
Found in Fabry disease; other lysosomal storage diseases.[31][Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].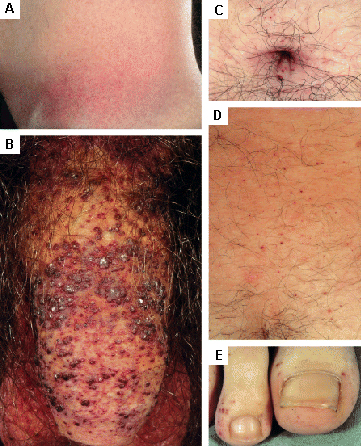 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry's disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].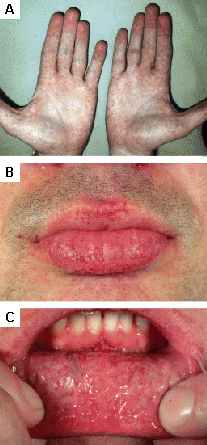
macular 'cherry red spot' on ophthalmoscopy
The choroid viewed through the fovea centralis appears as a red circular area surrounded by grey-white retina. Classical finding in infantile Tay-Sachs disease.[56]
optic atrophy or retinitis pigmentosa on ophthalmoscopy
Optic atrophy or retinitis pigmentosa are seen in juvenile form of Tay-Sachs disease.
corneal clouding on ophthalmoscopy
Other diagnostic factors
common
neurodevelopmental delay
hearing impairment/sudden deafness
Found in Fabry disease, mucopolysaccharidosis disorders.[49]
cataract on ophthalmoscopy
Characteristic cataract seen in Fabry disease with tortuous retinal vessels. Cataracts are also seen in Gaucher disease, mucopolysaccharidosis disorders, and other lysosomal storage diseases.[56]
eye movement disorder
progressive dementia and ataxia or gait disturbance
Seen in several juvenile and adult-onset lysosomal storage diseases, often accompanied by ataxia or gait disturbance (e.g., juvenile and chronic Tay-Sachs disease, Niemann-Pick type C).[54]
failure to thrive
joint contracture
depression
Very common in adults with Fabry disease.[12] Also found in Gaucher, Pompe, and Tay-Sachs diseases.
skeletal abnormalities including spinal gibbus
Frequent early sign in mucopolysaccharidosis disorders.[49]
hydrocephalus
Frequent early sign in mucopolysaccharidosis disorders.[49]
history of recurrent respiratory tract infections
psychosis
uncommon
premature stroke/transient ischaemic attack
Seen in Fabry disease (frequently posterior circulation).[31]
Risk factors
strong
male sex (mucopolysaccharidosis [MPS] II, Fabry disease)
Ashkenazi ethnicity
Carrier rate among Ashkenazi Jews is about 1 in 15 for Gaucher disease, 1 in 30 for Tay-Sachs, and 1 in 80 to 1 in 100 for Niemann-Pick type A.[11]
Use of this content is subject to our disclaimer