Etiology
Hirschsprung disease is a genetic disorder with a complex pattern of inheritance.[19][24] Genetic studies have identified variants in more than 24 genes associated with the disease.[1][26]
Rearranged during transfection (RET) is the most common gene implicated in Hirschsprung disease, with mutations found in 50% of familial cases and 15% to 35% of sporadic cases.[19][27][28] Mutations in RET are also implicated in multiple endocrine neoplasia type 2 (MEN2), which is also associated with Hirschsprung disease.[29] See Multiple endocrine neoplasia syndromes.
Endothelin receptor type B (EDNRB) is the second most commonly mutated gene in sporadic Hirschsprung disease, with a frequency of approximately 5%.[24] Mutations in EDNRB lead to Shah-Waardenburg syndrome which is characterized by congenital deafness, pigmentation abnormalities, and Hirschsprung disease.[30]
Other genes implicated in Hirschsprung disease include SOX10 and PHOX2B, which is associated with Haddad syndrome; a combination of Hirschsprung disease and congenital central hypoventilation syndrome.[31]
Pathophysiology
The absence of ganglion cells and the presence of hypertrophic nerves, as well as an increase in the enzyme acetylcholinesterase, are the keys to pathologic diagnosis of the dysfunctional bowel segment.[32][33][34][35][Figure caption and citation for the preceding image starts]: Histologic section including mucosa with submucosa of the rectum showing clusters of ganglion cells in the submucosal plexus. This excludes Hirschsprung disease at this levelCorman ML. Colon and rectal surgery. 5th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; used with permission [Citation ends].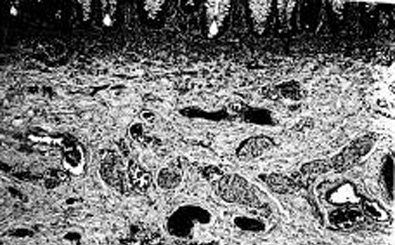 [Figure caption and citation for the preceding image starts]: Histologic section including mucosa and submucosa of the rectum showing tortuous and hypertrophic nerve trunks of the submucosal plexus. There is no evidence of any ganglion cell present. This establishes the diagnosis of Hirschsprung diseaseCorman ML. Colon and rectal surgery. 5th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Histologic section including mucosa and submucosa of the rectum showing tortuous and hypertrophic nerve trunks of the submucosal plexus. There is no evidence of any ganglion cell present. This establishes the diagnosis of Hirschsprung diseaseCorman ML. Colon and rectal surgery. 5th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2005:555; used with permission [Citation ends].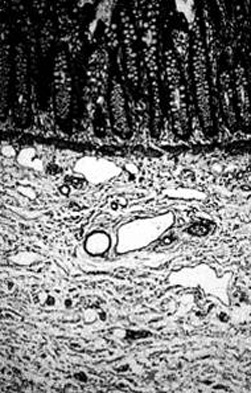
Pathology staining demonstrates a significant increase in the number of oversized nerve fibers located in the muscularis mucosa, the lamina propria, and the submucosa, and an increase in acetylcholinesterase activity. Hypertrophic nerves are those greater than 40 microns in diameter.[35]
Hirschsprung disease is believed to be caused by abnormal development of the enteric nervous system. During embryogenesis, there appears to be an arrest in the craniocaudal migration of the neuro enteric ganglion cells from the neural crest into the upper gastrointestinal tract, down through the vagal fibers, and along the distal intestine.[36] This is thought to result from mutations in genes such as RET, GDNF, EDNRB, SOX10, and PHOX2B, which are integral to the signaling pathways that regulate the migration of these cells during embryonic development.[1][37] As a consequence, ganglion cells are missing from the Auerbach myenteric plexus, the Henle plexus, and at the Meissner plexus.[18] The number and severity of mutations, in addition to epigenetic and environmental factors, are believed to influence the length of the aganglionic segment.[1]
Under normal circumstances, the ganglia appear to act as a final common path for both sympathetic and parasympathetic influences. Their absence may perhaps produce the uncoordinated contractions of the affected bowel. Spasm, lack of propulsive peristalsis, and mass contraction of the aganglionic segment have all been well documented, in addition to the lack of relaxation of the bowel and the spasm of the internal sphincter.[38][39] The clinical results of these pathophysiologic events is partial or total colonic functional obstruction.
The role of nitric oxide as a neurotransmitter responsible for the inhibitory action of the intrinsic enteric nerves is being elucidated.[40][41]
Classification
Length of aganglionic segment
Hirschsprung disease can be categorized by the length of the aganglionic segment. This determines the severity of the disease and subsequent management.[4]
Short-segment (rectosigmoid)
The aganglionic segment includes the rectum and much of the sigmoid colon. This comprises 80% to 85% of cases.[5]
Long-segment
The aganglionic segment extends beyond the sigmoid-descending colon (although this definition varies between studies).[4][6] People with long-segment disease often have more complicated courses and require particularly careful consideration with regard to treatment.[4][7] Many children with long-segment disease will require intestinal diversion as part of their initial management.[1] This comprises up to 20% of cases.[5]
Total colonic aganglionosis (TCA)
A very serious condition in which the entire colon is aganglionic, frequently including a variable length of terminal ileum. This comprises about 8% of cases and will usually require intestinal diversion followed by proctocolectomy and reconstruction later in life.[6][8]
Total intestinal
Total intestinal aganglionosis is rare (<1% of cases) and occurs when the total amount of ganglionated small bowel is less than 40 cm.[9] Treatment includes intestinal rehabilitation, parental nutrition, and sometimes intestinal transplantation.[10]
Ultrashort-segment
There is some debate about the existence of this subtype and a 2021 systematic review by the American Pediatric Surgical Association Outcomes and Evidence-Based Practice Committee recommended that this term should not be used.[4]
Use of this content is subject to our disclaimer