Turner syndrome occurs equally in all races and regions of the world. It is sporadic in occurrence except for rare cases in which a small X chromosome deletion may be passed from mother to daughter.
The number of females diagnosed prenatally may increase with increased use of noninvasive prenatal testing (NIPT).[6]Wang Y, Li S, Wang W, et al. Cell-free DNA screening for sex chromosome aneuploidies by non-invasive prenatal testing in maternal plasma. Mol Cytogenet. 2020;13:10.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7068885
http://www.ncbi.nlm.nih.gov/pubmed/32190123?tool=bestpractice.com
Regardless, a postpartum karyotype is necessary to confirm the diagnosis.[3]Gravholt CH, Andersen NH, Conway GS; International Turner Syndrome Consensus Group. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol. 2017;177:G1-G70.
http://www.eje-online.org/content/177/3/G1.long
http://www.ncbi.nlm.nih.gov/pubmed/28705803?tool=bestpractice.com
Historically, less than 10% of cases are diagnosed prenatally; a further 20% are detected in infancy owing to the presence of lymphedema, neck webbing, or congenital heart defects. Relatively few patients are diagnosed during early childhood and they usually present with short stature. The largest proportion of detection is at ages 10 to 16 years, owing to a combination of marked short stature and delayed puberty. Another 10% are diagnosed in adulthood, owing to secondary amenorrhea. There are similar ascertainment profiles for the US and Europe.[7]Stochholm K, Juul S, Juel K, et al. Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006;91:3897-3902.
http://jcem.endojournals.org/content/91/10/3897.full
http://www.ncbi.nlm.nih.gov/pubmed/16849410?tool=bestpractice.com
[8]McCarthy K, Bondy CA. Turner syndrome in childhood and adolescence. Expert Rev Endocrinol Metab. 2008;3:771-775.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2752892
http://www.ncbi.nlm.nih.gov/pubmed/19789718?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Ascertainment profile data of Turner syndrome; 300 females; Age DX, age (in years) at diagnosisFrom the personal collection of Carolyn Bondy, MS, MD (National Institute of Child Health and Human Development natural history study 2001-2007; McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775) [Citation ends].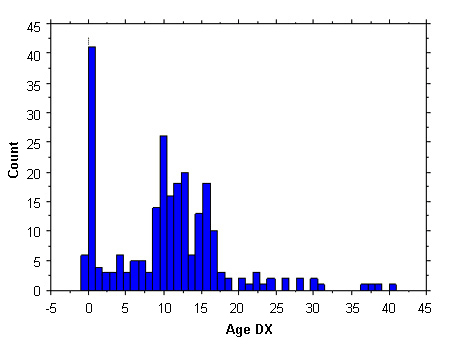
Whereas large-scale cytogenetic screening of newborns conducted in the 1970s and 1980s indicated a Turner syndrome incidence of about 1 case per 2000-2500 live female births,[9]Jacobs PA, Melville M, Ratcliffe S, et al. A cytogenetic survey of 11,680 newborn infants. Ann Hum Genet. 1974;37:359-376.
http://www.ncbi.nlm.nih.gov/pubmed/4277977?tool=bestpractice.com
[10]Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet. 1991;87:81-83.
http://www.ncbi.nlm.nih.gov/pubmed/2037286?tool=bestpractice.com
more recent surveys suggest a lower incidence of about 1 case per 5000 live female births due to prenatal diagnosis associated with terminations of pregnancies.[11]Iyer NP, Tucker DF, Roberts SH, et al. Outcome of fetuses with Turner syndrome: a 10-year congenital anomaly register based study. J Matern Fetal Neonatal Med. 2012;25:68-73.
http://www.ncbi.nlm.nih.gov/pubmed/21463211?tool=bestpractice.com