Etiology
There are no known predisposing or causative factors for Turner syndrome. It is sporadic in occurrence except for rare cases in which a small X chromosome deletion may be passed from mother to daughter.
Loss of all or a part of a sex chromosome, X or Y, during early embryogenesis usually results in the development of a female fetus with developmental anomalies. Monosomy for the Y-chromosome is nonviable. Those 46,XY fetuses that lose a Y chromosome relatively late in development may have enough Y chromosome input to drive partial or complete male development. However, these patients are identified as males, and are considered under the diagnosis of "disorders or differences of sex development" rather than Turner syndrome. Males with 45,X cell lines have variable degrees of short stature, delayed puberty or infertility, and risk for cardiovascular disorder.[12]
The phenotype is determined by the timing of chromosomal loss, persistence of normal cell lines (relative abundance and tissue distribution), and the genetic background.
Pathophysiology
Homologous regions of the sex chromosomes (pseudoautosomal region [PAR]) behave like autosomes in that they undergo homologous pairing and recombination, and contain genes that escape X-inactivation. These genes are presumably required in dual dosage in both males and females. There are about 20 annotated genes and several other predicted genes in PAR1, located at the Xp terminus. A smaller set of homologous genes is found in PAR2, at the Xq terminus. Haploinsufficiency for PAR1 genes is implicated in Turner syndrome.
[Figure caption and citation for the preceding image starts]: Pseudoautosomal regions of X and Y chromosomesFrom the personal collection of Carolyn Bondy, MS, MD [Citation ends].
The specific pathophysiology behind the developmental anomalies seen in Turner syndrome is poorly understood.
Impaired long-bone growth and skeletal development leading to short stature has been linked with haploinsufficiency for a specific sex chromosome gene, SHOX, located in the PAR, which encodes a transcription factor expressed in developing bone and cartilage.[13][14]
Haploinsufficiency for other pseudoautosomal genes causes the congenital cardiovascular defects that produce high mortality in Turner syndrome.[15][16] Two genes,TIMP1 and TIMP3, which are both located on the short arm of the X chromosome when hemizygous increase the risk of aortopathy with an odds ratio of 12.86.[17]
The cause of premature ovarian failure remains unclear, although genes located on both short and long arms of the X chromosome have been implicated.[18] The X-inactivation center (XIC) is located on the long arm, Xq. Deletion of Xq distal to the XIC is associated with premature ovarian failure more than other Turner syndrome features. The ovaries form normally in 45,X female fetuses, although most demonstrate accelerated oocyte death and ovarian degeneration into fibrous streaks.[19]
Haploinsufficiency for genes located on Xp results in distinctive neurocognitive traits, such as higher verbal versus visual-spatial performance scores and difficulties in recognition of facial expressions.[20]
Classification
Cytogenetic classification[4]
Nonmosaic 45,X
Single X chromosome in all somatic cells (monosomy X); comprises approximately 60% of patients with Turner syndrome.
Fragmented X or Y
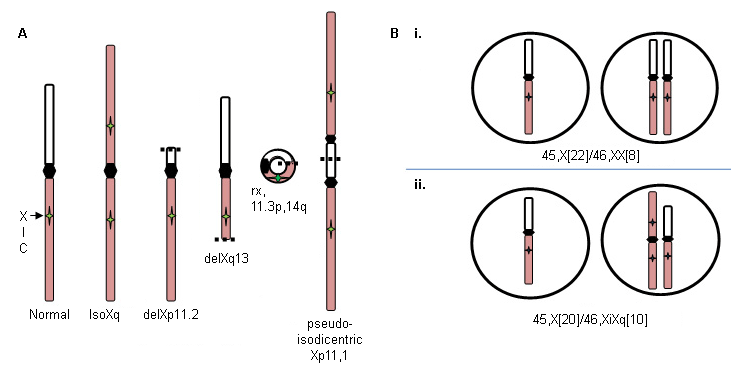
Xp deletions (46,X,delXp), isoXq chromosomes (46,X,iXq), Xq deletions, and ring X or Y chromosomes with substantial interstitial deletions. The isoXq chromosome, consisting of deletion of Xp and fusion of 2 long arms, is the most common structural anomaly associated with Turner syndrome. Deletion of major portion of Xp is also associated with the syndrome. Often, the abnormal sex chromosome is lost in some cells during embryonic development, resulting in a mosaicism of 45,X cell line in addition to the 46,X, fragX line (see figure A below).
Mosaic 45,X
There are two types of mosaicism.
Loss of a sex chromosome during early embryonic cell divisions may result in a mixture of normal 46,XX cells and 45,X cells in variable proportions throughout body tissues, and includes approximately 15% of patients with Turner syndrome: for example, 45,X (50%)/46,XX (50%) (see figure B i. below). The relative proportion of normal cells in different tissues will influence the phenotype.
By contrast, formation of a 46,X,abnX embryo is often accompanied by loss of the fragmented X during some embryonic cell divisions, resulting in mosaicism for a monosomic, 45,X cell line along with a cell line containing a fragmented X; for example, iXq (see figure B ii. below). This individual has no normal cells, and the full phenotype is expected. [Figure caption and citation for the preceding image starts]: X chromosome abnormalities in Turner syndrome; see text for explanationFrom the personal collection of Carolyn Bondy, MS, MD [Citation ends].
Use of this content is subject to our disclaimer