History and exam
Key diagnostic factors
common
Family history
These are inherited disorders and should be assessed after appropriate consultation with a clinical geneticist. Occasionally patients do not have a family history, either because the families are small (these are mainly autosomal-recessive disorders and carriers are generally asymptomatic), or because it may be a new mutation. Family history in the extended pedigree is common in X-linked lysosomal storage diseases (Fabry, mucopolysaccharidosis [MPS] II, and Danon).
onset in childhood (MPS, Pompe, Gaucher, Fabry, Niemann-Pick type A)
Age <1 year: the severe forms of the mucopolysaccharidosis (MPS) disorders, classic Pompe disease, and neuronopathic forms of Gaucher disease present in early infancy.[30][35][46][50][80] Early age of onset is a strong indicator of a severe long-term course, and is often associated with low residual enzyme activity.
Age 1 to 10 years: most MPS disorders (including attenuated variants) and Niemann Pick type A will present by this age.[81] Most males with classic Fabry disease will have had some symptoms, although the average age of diagnosis in males is older.[38]
onset in adolescence (Fabry, Pompe, Gaucher types 1, 3, mucopolysaccharidosis, Niemann-Pick types B, C)
Age 10 to 20 years: most type 3 Gaucher disease; most of the more severe type 1 Gaucher disease but not all; most classic Fabry disease; and most Pompe, including adult onset, will be symptomatic, although not all diagnosed at these ages. Niemann-Pick types B and C are variable; severe phenotypes present earlier, milder phenotypes present later. Attenuated forms of mucopolysaccharidosis disorders are also highly variable.
onset in adulthood (Fabry, Gaucher type 1, Pompe)
Age 20 to 50 years: typical age of onset for most patients with type 1 Gaucher disease, and the age that patients with classic Fabry disease develop clinical problems.
Age >50 years: lysosomal storage diseases are unlikely to present in older age; exceptions are milder forms of type 1 Gaucher disease, atypical Fabry disease, and some patients with milder Pompe disease.
hepatomegaly and/or splenomegaly
Hepatomegaly is seen in type 2 and 3 Gaucher, mucopolysaccharidosis disorders, Pompe, and Niemann-Pick diseases.[5][30][46][49][54]
Splenomegaly is seen in type 2 and 3 Gaucher, mucopolysaccharidosis disorders, and Niemann-Pick disease.[5][30][46][49][54]
Hepatosplenomegaly is common in mucopolysaccharidosis disorders, Gaucher disease, and Niemann-Pick types A, B, and C.[5][30][46][49][54]
hyperacusis
Characteristic of Tay-Sachs disease.[82]
history of renal failure
Found in adult Fabry disease.[31]
skin rash/cutaneous lesions
Found in Fabry disease; other lysosomal storage diseases.[31][Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry disease: (A) flank, (B) genitals, (C) umbilicus, (D) lower back, (E) toesOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].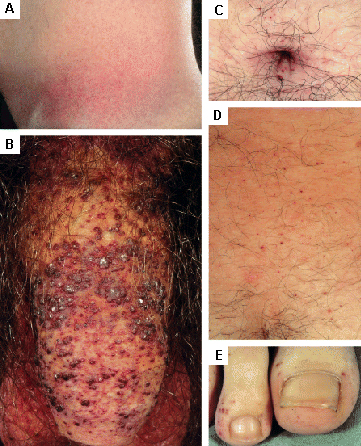 [Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Cutaneous lesions in Fabry disease: (A) palms, (B) lips, (C) labial mucosaOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; used with permission [Citation ends].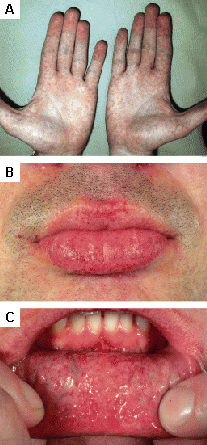
macular "cherry red spot" on ophthalmoscopy
The choroid viewed through the fovea centralis appears as a red circular area surrounded by gray-white retina. Classical finding in infantile Tay-Sachs disease.[56]
optic atrophy or retinitis pigmentosa on ophthalmoscopy
Optic atrophy or retinitis pigmentosa are seen in juvenile form of Tay-Sachs disease.
corneal clouding on ophthalmoscopy
Other diagnostic factors
common
neurodevelopmental delay
hearing impairment/sudden deafness
Found in Fabry disease, mucopolysaccharidosis disorders.[49]
cataract on ophthalmoscopy
Characteristic cataract seen in Fabry disease with tortuous retinal vessels. Cataracts are also seen in Gaucher disease, mucopolysaccharidosis disorders, and other lysosomal storage diseases.[56]
eye movement disorder
progressive dementia and ataxia or gait disturbance
Seen in several juvenile and adult-onset lysosomal storage diseases, often accompanied by ataxia or gait disturbance (e.g., juvenile and chronic Tay-Sachs disease, Niemann-Pick type C).[54]
failure to thrive
joint contracture
depression
Very common in adults with Fabry disease.[12] Also found in Gaucher, Pompe, and Tay-Sachs diseases.
skeletal abnormalities including spinal gibbus
Frequent early sign in mucopolysaccharidosis disorders.[49]
hydrocephalus
Frequent early sign in mucopolysaccharidosis disorders.[49]
history of recurrent respiratory tract infections
psychosis
uncommon
premature stroke/transient ischemic attack
Seen in Fabry disease (frequently posterior circulation).[31]
Risk factors
strong
male sex (mucopolysaccharidosis [MPS] II, Fabry disease)
Ashkenazi ethnicity
Carrier rate among Ashkenazi Jews is about 1 in 15 for Gaucher disease, 1 in 30 for Tay-Sachs, and 1 in 80 to 1 in 100 for Niemann-Pick type A.[11]
Use of this content is subject to our disclaimer