Aetiology
Wilms' tumour is a genetically heterogeneous neoplasm.[3][14] It may be inherited or occur sporadically. The cancer genes that underpin Wilms' tumour are diverse and involve around 40 genes.[3] Most genes act in gene expression control and growth factor signalling.[3] Germline mutations are present in at least 10% of patients and include the first gene implicated in Wilms' tumourigenesis, the WT1 (Wilms' tumour 1) gene.[15] Other germline mutations detected in the Children's Oncology Group TARGET initiative paper include TP53 (tumour protein p53; 17p13), DICER1 (dicer 1, ribonuclease III; 14q32), DIS3L2 (DIS3 like 3'-5' exoribonuclease 2; 2q37), and some unexpected germline variants involving PALB2 (partner and localiser of BRCA2; 16p12) and CHEK2 (checkpoint kinase 2; 22q12).[15] The most prevalent somatic mutations are in the WT1 (11p13), CTNNB1 (catenin beta 1; 3p22), AMER1 (APC membrane recruitment protein 1; Xq11), IGF2 (insulin-like growth factor 2; 11p15), TP53 (17p13), MYCN (MYCN proto-oncogene, bHLH transcription factor; 2p24), SIX1 (SIX homeobox 1; 14q23) and SIX2 (SIX homeobox 2; 2p21), and microRNA processing genes.[14]
The risk for developing Wilms' tumour is increased in children with congenital anomalies. In one study of childhood cancer in Great Britain, congenital anomalies were found in around 8% of children with Wilms' tumour.[16] The risk is increased among children with certain congenital overgrowth syndromes, such as Perlman's syndrome, Beckwith-Wiedemann syndrome, and Simpson-Golabi-Behmel syndrome, and also among children with certain congenital non-overgrowth syndromes, such as Denys-Drash syndrome and WAGR (Wilms' tumour, aniridia, genitourinary abnormalities, range of developmental delays) syndrome.[3][17]
Children with congenital urogenital anomalies such as hypospadias, atypical genitalia, fused (horseshoe) kidney, or cryptorchidism are also predisposed to developing Wilms' tumour.[3][16][18]
Studies have investigated the association between Wilms' tumour and antenatal exposure to harmful environmental factors or maternal lifestyle risk factors; most studies are inconclusive or require further investigation. However, a meta-analysis of case-control studies identified a link between parental pesticide exposure during the preconception or pregnancy period and an increased risk for Wilms' tumour.[19]
Pathophysiology
Many different genetic changes converge into a limited number of developmental pathways resulting in Wilms' tumour oncogenesis.[15] By understanding these identified pathways, potential targeted therapeutic approaches can be applied in a clinical setting.[3]
Wilms' tumour oncogenesis is thought to be a multi-step process that arises from aberrant fetal nephrogenesis, with many Wilms' tumour-related genes having pivotal roles in the developing kidney.[3][14] As the differentiation arrest of renal progenitor cells is incomplete, all three lineages of the developing kidney (blastema, epithelia, and stroma) can be identified in classic triphasic Wilms' tumour histology.[14]
Nephrogenic rests are microscopic foci of primitive blastemal renal elements found in normal kidney tissue in an intralobar or perilobar location, and are derived from renal stem cells. Nephrogenic rests are found in >90% of patients with bilateral Wilms' tumours and in around 40% of patients with unilateral sporadic Wilms' tumour, and are considered precursor lesions to Wilms' tumour.[3] The acquisition of additional genetic and epigenetic changes then leads to tumour formation.
The first genetic model, or 'two-hit hypothesis' (two rate-limiting mutational events that occur sequentially), postulates that children who develop hereditary Wilms' tumour are born with a constitutional pre-zygotic DNA mutation in one allele of a gene, which is the initiating event, and that this event is followed by another genetic event in the same or different gene locus, leading to formation of the tumour.[20][21] The WT1 gene (11p13) and the WT2 gene (11p15) are tumour suppressor genes that are known to play an important role in the normal development of the urogenital tract.[20] Inactivation of these genes appears to be an early genetic event in the evolution of Wilms' tumour. Loss of heterozygosity (LOH) at 1p and 16q is thought to be a secondary event that ultimately results in Wilms' tumour formation.[20]
Studies suggest that aberrant expression of the IGF-2 oncogene, resulting from the loss of imprinting of the maternal IGF-2 gene, may be one of the most common epigenetic alterations in Wilms' tumour.[22] Furthermore, intralobar nephrogenic rests are more likely to be associated with the identical genetic mutation found in the Wilms' tumours originating in the same host.[23]
Altogether, these studies suggest that both nephrogenic rests and the tumour may be clonally derived from a common embryonic renal stem cell and that multiple sequential genetic and epigenetic events such as inactivation of tumour suppressor genes and LOH at 1p and 16q may occur within these lesions leading to tumourigenesis.[24] An analysis of histologically normal tissue also supports the hypothesis that clonal expansions within normal kidney tissue, some of which harbour driver variants, antedate the development of Wilms' tumour.[25]
Copy number gains at the chromosome region 1q21.1-q31.3 have been shown to be significantly associated with relapse, as well as allelic imbalances at chromosome arms 1p, 1q, 3p, 3q, and 14q.[26][27]
Analysis of primary-relapse tumour pairs (where DNA is available from the primary tumour to compare with the relapse tumour) suggests that WT2 (11p15) LOH (and other copy number changes) and mutations in WT1 and MLLT1 typically occur early, but mutations in SIX1, MYCN, and WTX are late developments.[28]
[Figure caption and citation for the preceding image starts]: Chromosome 11 deletionNational Cancer Institute; used with permission [Citation ends].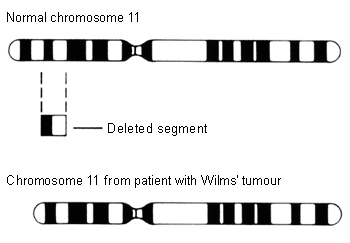
Use of this content is subject to our disclaimer