Approach
Timely diagnosis of CP allows instigation of appropriate management and supportive services.[69][70] Diagnosis is primarily clinical, supported by imaging.[56] Examination includes assessment of active and passive range of motion, motor power, selective voluntary motor control, muscle tone, and sensation of the limbs. Further testing is individualised based on the history and examination findings.[71]
Progressive brain deterioration is not a feature of CP and should be investigated for an alternative diagnosis.
History
A thorough history should be undertaken for antenatal risk factors (teratogen exposure, maternal illness, chorioamnionitis, antenatally diagnosed fetal brain malformations), perinatal risk factors (instrumental delivery, non-vertex presentation, placental abruption, rupture of the uterus, prolonged/obstructed labour, postmaturity), and postnatal risk factors (hyperbilirubinaemia, neonatal sepsis, respiratory distress, early-onset meningitis, intraventricular haemorrhage, head injuries prior to age 3 years). Around 25% of infants who survive neonatal seizures develop CP.[35]
Family history may point to a genetic or a familial metabolic aetiology. Low income levels were associated with a twofold increase in CP in one study.[17] A social history may elicit any concerns regarding head injury due to child abuse or shaken baby syndrome.
Developmental milestones
A full history of all developmental milestones must be obtained. Developmental examination, including screening, can be done with the help of tools such as Denver II and Bayley III (Denver Prescreening Developmental Questionnaire and Bayley Scales of Infant Development, respectively). Special motor assessment tools during the first year of life for infants born preterm help detect changes in motor quality.[72] Assessment of potential delay of developmental milestones should use corrected age for children born preterm, aged up to 2 years.[56][73]
Delayed motor milestones are often the key diagnostic factor. Typically, children sit by age 6 months, crawl with reciprocal locomotion by 9 months, walk between 12 and 18 months, talk in short sentences by 2 years, and manipulate stairs in an adult fashion (step over step) by 3 years.
A delay in speech development may reflect motor delay or an intellectual disability. Speech delay (observed in >50% of patients with CP) is more prevalent in children with total body involvement.
Cognitive impairment is observed in around 50% of patients with CP, and severe cognitive impairment in around 25%.[56]
Spastic CP
Spastic CP is the most common subgroup, and is characterised by a velocity-dependent increase in tonic stretch reflexes, with exaggerated tendon jerks and clonus resulting from hyperexcitability of the stretch reflex.[3] The severity varies from mild involvement of a single limb to severe total body involvement.
Overall diagnostic principles involve confirming the diagnosis when obvious and deferring the diagnosis when equivocal, while continuing to treat the symptoms. Approximately half of children outgrow a tentative diagnosis of spastic CP, established at age 1 year, by age 7 years.[74] Spasticity typically remains stable after age 5 years.
Most children achieve 90% of their motor potential by age 5 years, and the likelihood of a child starting to walk after age 7 years is small.[75][76] Some paediatricians believe that a definitive diagnosis of spastic CP should not be made until age 5 years to avoid overdiagnosis.
Immediate postnatal period
Characterised by diminished muscle tone, which becomes progressively hypertonic at ages 16 to 18 months.
Early infancy
Diagnosis of CP can be made before age 5 months (corrected age for children born preterm) with up to 98% sensitivity using Prechtl's Assessment of General Movements (GMs), the Hammersmith Infant Neurological Examination (HINE), and neuroimaging.[77][78][79]
After age 5 months (corrected), use of the HINE and the Developmental Assessment of Young Children (DAYC) 2 is recommended in addition to neuroimaging.[69][70][80][81]
Other factors to assess infants include the presence of sustained clonus or other persistent pathological reflexes.[69] Sustained clonus is defined as clonus for more than 3 beats at a time, usually at the ankle level. Reflexes and reactions that are poor prognostic factors for the development of independent walking include:
Retention of asymmetric and symmetric tonic neck reflexes
Retention of Moro (startle) reflex
Retention of neck righting reflex
Presence of lower-extremity extensor thrust response
Lack of parachute reaction
Lack of foot placement reaction.[82]
Early childhood
Spasticity typically does not develop until the second year of life when the child attempts activities. It is confirmed by hypertonia (velocity-dependent resistance to passive motion), abnormally increased deep tendon reflexes, and the presence of clonus. Spasticity is graded by the modified Ashworth scale.[83] It may be accompanied by a 'clasp knife' phenomenon, which refers to resistance to passive motion that abruptly decreases, letting the limb move more easily.
Patients also manifest selective voluntary motor control impairment, with an inability to move isolated joints without obligatory movement of non-agonist joints. This can be assessed by tools such as the selective control assessment of the lower extremity (SCALE).[84]
Patients with spastic diplegia have bilateral involvement, with the lower extremities being more involved than the upper extremities. Patients with spastic quadriplegia have significant involvement of all four extremities and higher rates of intellectual, oral-motor, and visual impairment.
Patients manifest varying degrees of ambulatory ability based on the gross motor function classification system (GMFCS). CanChild: Gross Motor Function Classification System - Expanded & Revised (GMFCS - E&R) Opens in new window
Mid-childhood
The effects of spasticity on the musculoskeletal system may result in progressive contractures or deformity, especially during periods of rapid growth, and can develop by age 5 years. Fixed contractures are not altered by sleep or anaesthesia. Evaluations for hip subluxation and scoliosis should be ongoing. [Figure caption and citation for the preceding image starts]: Dislocated hip in a patient with CPFrom the collection of William L. Oppenheim; used with permission [Citation ends].
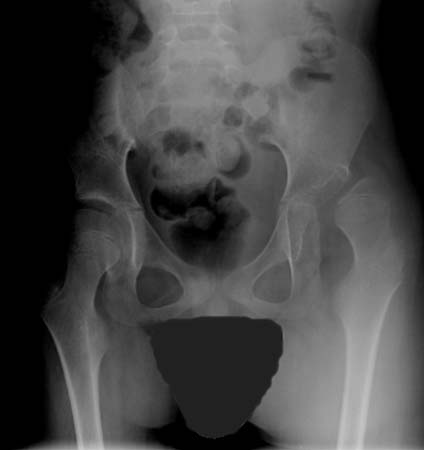
The severity of contracture and deformity tends to be less in children with hemiplegia than in those with more global involvement; hip dysplasia and deformity are rare in hemiplegia, but should not be ignored.[85]
Dyskinetic CP
Dyskinetic CP includes movement disorders originating in the extra-pyramidal system, leading to dystonia, chorea, and athetosis. Patients present with involuntary, recurring, and occasionally stereotyped movements with a varying muscle tone. Athetosis and chorea present in early childhood, while dystonia presents in later childhood or adolescence. Although physical impairment may be severe in these patients, preservation of intellectual ability is common.
Dystonia is characterised by involuntary, sustained contractions resulting in twisting and abnormal postures. Patients may have associated rigidity, described as a constant resistance or 'lead pipe' in character, which does not change with velocity.
Chorea presents as rapid, involuntary, jerky, and fragmented motions. Tone is usually decreased but fluctuating.
Athetosis is characterised by slower, constantly changing, writhing, or contorting movements.
Ataxic CP
Ataxic CP involves loss of muscular coordination with abnormal force and rhythm, and impairment of accuracy, resulting in gait and truncal ataxia, poor balance, past pointing, terminal intention tremor, scanning speech, nystagmus and other abnormal eye movements, and hypotonia.
Proximal musculature of the pelvis, scapula, and trunk are particularly affected, resulting in the patient compensating distally for the proximal impairment. An alternative diagnosis should be pursued in patients with pure ataxia, as this is rare in CP.
Mixed CP
Many patients with CP exhibit more than one type of movement disorder (e.g., presence of a dyskinetic movement disorder along with spasticity, rigidity, or postural hypertonia). In these patients, the dyskinetic component may not manifest until after the spasticity is apparent.
Gait
An abnormal gait can be detected after walking age, which is frequently delayed. Gait evaluation may be helpful in diagnosing the subtype of CP.
Spastic CP
Abnormalities include asymmetry, toe walking, limb rotational deviations, 'scissoring' (crossing of the legs), poor balance, excessive joint flexion, foot drag, and flexed arm posture.
Hemiplegia: excessive plantar flexion may manifest as unilateral toe-walking in the young child or knee hyperextension in the older child or adult. Patients with spastic hemiplegia are typically ambulatory.
Diplegia: spasticity of the plantar flexors may cause bilateral toe-walking in the young child. However, this is rarely seen in the school-age child or in adults, who have a crouched gait due to weakness of plantar flexors with hip and knee flexion contractures. Hip adductor or medial hamstring spasticity may manifest itself as scissoring during upright activities. Internal rotation of the femora or tibiae may also mimic scissoring.
Quadriplegia: the majority of these patients are not able to walk for any substantial distance and require external support of assistive devices such as walkers and gait trainers.
Ataxic CP
Patients have an ataxic gait and suffer frequent falls. While ataxia may be the primary movement disorder, it typically occurs in concert with other movement disorders including spasticity and dyskinesia.
Dyskinetic CP
Patients with dyskinesia may or may not walk. They exhibit excessive involuntary movements that often interfere with stability and balance.
Investigations
Individualised, based on the history and examination findings.[71]
Neuroimaging
May reveal periventricular leukomalacia, congenital malformation, stroke, or haemorrhage. Cystic lesions may be present, and are not unusual in cases of hemiplegia as a result of antenatal or perinatal vascular accidents.[Figure caption and citation for the preceding image starts]: Periventricular leukomalacia. Figure A is a normal 14-year-old female: the curved arrow shows normal lateral ventricle and the straight arrow shows normal white matter. Figure B is a 14-year-old female with CP: the curved arrow shows enlarged ventricle and the straight arrow shows diminished volume of the white matter due to periventricular leukomalaciaCourtesy of Noriko Solomon, Assistant Professor of Radiology, David Geffen School of Medicine at UCLA, Los Angeles [Citation ends].

Magnetic resonance imaging (MRI) of the brain is the neuroimaging modality of choice and is abnormal in up to 80% of patients with CP, especially those with prematurity or other risk factors.[36][37] Every child with an equivocal diagnosis of CP should have an MRI of the brain.[86] Over 50% of children with CP were born after normal term pregnancies and may not routinely have had an MRI during the neonatal period. If the presentation is typical of CP, imaging can be delayed until the child is old enough to have the scan without sedation, typically between ages 5 and 7 years.[37]
Ultrasound and computed tomography of the brain are not as sensitive as MRI in detecting brain abnormalities, but may help in diagnosis and prognosis.[87]
Coagulation studies
May be abnormal. Should be considered in patients with hemiplegia who have a high incidence of single-hemisphere infarction.
Genetic evaluation
Indicated for patients with dysmorphic features such as abnormal skin creases, low-set ears, lack of a nasal bridge, and hypo- or hypertelorism, or those with a suspicion of familial disease.[71] Patients may also have multiple congenital anomalies such as congenital spine lesions, abnormal kidneys, or congenital contractures.
Features may be evident in the neonatal intensive care or may become evident later in childhood.
Metabolic screen
Consider in newborns with suspected inborn errors of metabolism, who typically present with seizures, unexplained acidosis, and coma.
Some metabolic diseases may come to light following routine blood screening.[88][89]
X-rays
Depending on the physical findings, serial x-rays are required from age 3 years (e.g., hip x-rays to monitor for subluxation of hips, and/or spinal x-rays to confirm progression in patients with scoliosis, kyphosis, or lordosis). [Figure caption and citation for the preceding image starts]: Dislocated hip in a patient with CPFrom the collection of William L. Oppenheim; used with permission [Citation ends].
Children with a GMFCS level of I or II need less monitoring, starting by age 3 years; those with level III should be monitored at least every other year during active growth; and children with level IV or V need assessment at least every year during active growth.[90] CanChild: Gross Motor Function Classification System - Expanded & Revised (GMFCS - E&R) Opens in new window
The standard for measuring hip subluxation on an anteroposterior view of the pelvis is known as the migration index. The Hipscreen.org site contains a useful smartphone application, which superimposes an adjustable template onto a photograph of the hip x-ray, automating the process.
Instrumented gait analysis
Gait laboratories offer kinematic and kinetic analysis of gait.
Standardised objective tools including computer-motion analysis, electromyography, and force plate recordings are used to identify and quantify features of movement patterns. These data are typically represented in comparison with a database of individuals without disability and provide detailed information on motion and joint forces that cannot be observed clinically. For example, intoeing may be due to torsional deformities of the tibia or femur, soft-tissue contractures about the hip, or variation in pelvic motions in the transverse plane.
Observational gait analysis and a detailed physical examination can be effective in many cases, but instrumented gait analysis usually can distinguish among the possible causes to direct surgical intervention.[91]
Assessment tool
Measures useful in the diagnosis and follow-up of patients with CP include:[92]
Prechtl's Assessment of General Movements (GMs)[77]
Hammersmith Infant Neurological Examination (HINE) (a neurological examination tool for evaluating children aged 2 to 24 months [corrected age for children born preterm])[78]
Developmental Assessment of Young Children (DAYC) 2[81]
Paediatric evaluation of disability index (a measure of global functioning)[93]
Gross motor performance measure[94]
Gross motor function classification system CanChild: Gross Motor Function Classification System - Expanded & Revised (GMFCS - E&R) Opens in new window
Manual ability classification system (MACS) Manual Ability Classification System Opens in new window[9]
Communication function classification system (CFCS) Communication Function Classification System Opens in new window[10]
Gross motor function measure[7][8] CanChild: GMFM score sheets Opens in new window
Physician's rating scale observational approach[95]
Modified Ashworth scale for spasticity[83]
Barry-Albright dystonia scale[96]
International classification of functioning disability and health (emphasising societal integration as the endpoint of health outcome) WHO: International classification of functioning, disability and health Opens in new window
Functional mobility scale[97]
Selective control assessment of the lower extremity[84]
Denver Prescreening Developmental Questionnaire (Denver II)
Bayley Scales of Infant Development (Bayley III)
Cerebral palsy computerised adaptive test (CP-CAT).[100]
Reviews have described upper-extremity assessment tools for patients with hemiplegic CP.[101][102]
Use of this content is subject to our disclaimer