History and exam
Key diagnostic factors
common
family history
If a family member with TSC is a first-degree relative (i.e., a parent), a 50% chance exists for the patient to have the disorder. This 50% risk remains when multiple siblings are affected, and assumes that one parent unknowingly has the disorder or that there is gonadal mosaicism in one of the parents. This is a condition whereby the only organ affected with the TSC mutation is the gonads and their affected gametes, allowing the potential for multiple affected children from an otherwise unaffected parent. The prevalence is estimated to be <2% of all parents with an affected child.[2][3][20]
However, the majority (around two-thirds) of people with TSC have sporadic occurrence.[4] A three-generation family history should be obtained to assess for additional family members at risk of TSC.[18]
epilepsy
The most common presenting symptom of the disorder, epilepsy, occurs in up to 60% to 90% of patients with TSC over a lifetime.[23]
All seizure types may develop, with the exception of pure absence epilepsy.
Epilepsy most often becomes manifest in childhood, and infantile spasms may be the presenting symptom in one third of patients.[2][3][24] Infantile Spasms Action Network: what does IS look like? Opens in new window
cardiac rhabdomyoma (single or multiple)
Affects approximately 60% of children and 20% of adults with TSC.[25] Among infants with multiple lesions, 80% or more are ultimately diagnosed with TSC. These lesions may be identified at 22 weeks of gestation. Most patients have a mean of 3 lesions, 3 to 25 mm in size but rarely larger. They are identified in the ventricles more often than the atria, and in the septum more often than the walls.
Symptoms (arrhythmias, outflow obstruction, thromboembolism) are rare and typically evident in the neonatal period. These lesions generally regress during early childhood without the need for intervention.[2][3][26][Figure caption and citation for the preceding image starts]: Cardiac rhabdomyoma on sagittal T1 MRICourtesy of Dr Francis J. DiMario Jr [Citation ends].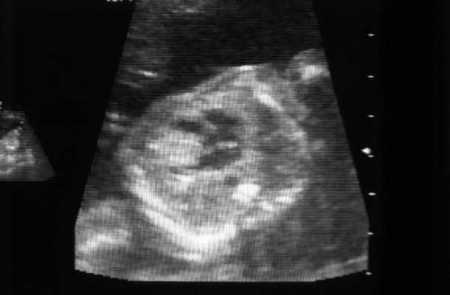
renal angiomyolipomas
One large-scale study found that renal angiomyolipomas (AMLs) are reported in nearly 50% of patients, and in patients with ongoing renal AML, 88% exhibited multiple lesions, 84% had bilateral lesions, 33% had lesions larger than 3 cm, and 21% had growing lesions.[28] One-quarter of patients also had renal cysts.[28]
AMLs increase in size and number over life, whereas cysts may regress. Multiple cysts are similar to polycystic kidney disease, with the cysts made up of hypertrophic eosinophilic epithelial cells. The AMLs are made of a benign mix of blood vessels, smooth muscle, and fat. Rarely, there can be a malignant AML or renal cell carcinoma.[28]
Most AMLs produce pain and haemorrhage in adulthood, whereas the cysts are more likely to cause hypertension and renal failure. Serious renal compromise occurs in <5% of patients overall.[2][3][26][Figure caption and citation for the preceding image starts]: Multiple renal angiomyolipomas on axial CTCourtesy of Dr Francis J. DiMario Jr [Citation ends].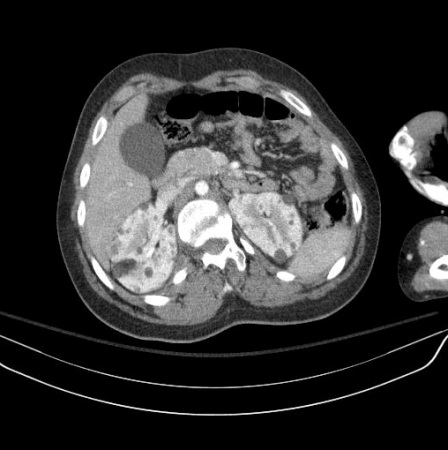 [Figure caption and citation for the preceding image starts]: Multiple renal angiomyolipomas on coronal T1 MRICourtesy of Dr Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Multiple renal angiomyolipomas on coronal T1 MRICourtesy of Dr Francis J. DiMario Jr [Citation ends].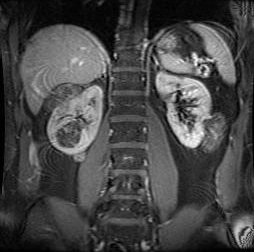
lymphangioleiomyomatosis of the lung
Affects up to 50% of women with TSC and appears to be hormonally sensitive.[29]
Symptomatic presentations can be with spontaneous pneumothoraces or chylothorax.
Lymphangioleiomyomatosis (LAM) produces progressive cystic cavitations within the lungs, which lead to declining air exchange and, ultimately, death. Treatment is supportive until lung transplantation can be provided.
This lung lesion in TSC is felt to be caused by metastases from underlying renal angiomyolipomas. Sporadic LAM also occurs in women without TSC.[2][3][26][Figure caption and citation for the preceding image starts]: Cystic lesions in lymphangioleiomyomatosis of the lung (LAM) on axial CTCourtesy of Dr Francis J. DiMario Jr [Citation ends].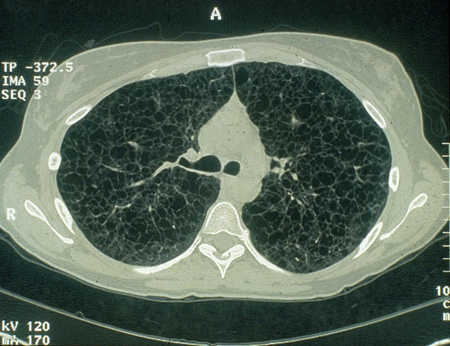
cerebral subependymal calcified nodules
Subependymal nodules are identifiable adjacent to the ventricular wall as small protrusions into the cerebrospinal fluid cavity.
These are often calcified by infancy, but this calcification may evolve more gradually over childhood. The characteristic calcification is readily identifiable with computed tomography, but can also be identified with magnetic resonance imaging.
The nodules are composed of dysplastic astrocytes and mixed lineage astrocytic components.[2][3][26][Figure caption and citation for the preceding image starts]: Subependymal calcified nodules on CTCourtesy of Dr Francis J. DiMario Jr [Citation ends].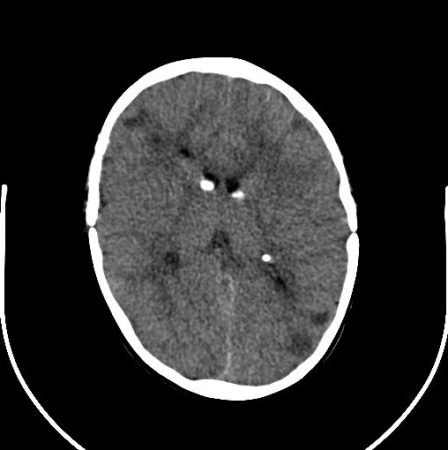 [Figure caption and citation for the preceding image starts]: Subependymal nodules and cortical tubers on axial T2 MRICourtesy of Dr Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Subependymal nodules and cortical tubers on axial T2 MRICourtesy of Dr Francis J. DiMario Jr [Citation ends].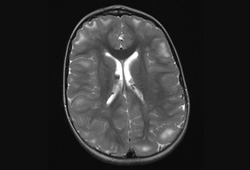
multiple cortical tubers and/or radial migration lines
Regions of focal cortical dysplasia extending into the juxtacortical areas of white matter giving the appearance of gyral mass lesions. They can be identified as low-density areas on computed tomography and high-intensity areas on T2/fluid attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) sequences.
Migration lines are seen as linear tracks of high-signal-intensity lesions on T2 MRI sequences extending centripetally perpendicular to the ventricular margins that represent a trail of incompletely migrated cellular components along radial glia remnants.
giant cell astrocytoma
Subependymal nodules, with a predilection for the region around the foramen of Monro, may enlarge due to cellular proliferation. Thus, by virtue of size and location, they are designated a subependymal giant cell astrocytoma (SEGA).
These occur in as many as 10% to 15% of patients, usually by 10 years of age.[30]
Enlargement may produce obstruction of cerebrospinal fluid pathways and invasion into the underlying hypothalamic and chiasmatic region. It is not possible to predict the rate of growth and impending need for neurosurgical intervention. Follow-up imaging and clinical judgement are required.[2][3][26][Figure caption and citation for the preceding image starts]: Large subependymal giant cell astrocytoma on MRI (A-axial T2)Courtesy of Dr Francis J. DiMario Jr [Citation ends].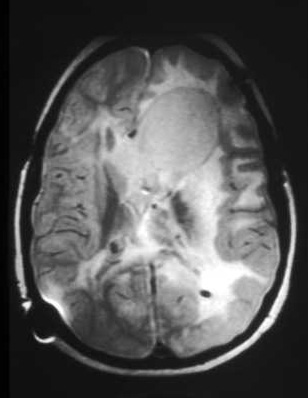 [Figure caption and citation for the preceding image starts]: Large subependymal giant cell astrocytoma on MRI (B-sagittal T1)Courtesy of Dr Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Large subependymal giant cell astrocytoma on MRI (B-sagittal T1)Courtesy of Dr Francis J. DiMario Jr [Citation ends].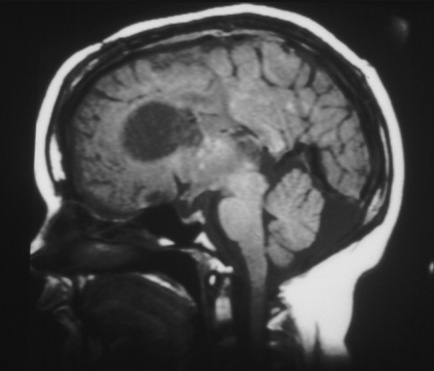
facial angiofibromas
These hamartomatous nodules of vascular and connective tissue distribute in a butterfly pattern. They most commonly appear at about 3 to 5 years of age, increase in size and number over life, and are identified in up to 88% of patients with TSC.[31] They can also be seen in about 80% of patients with MEN-1 (multiple endocrine neoplasia).[32][Figure caption and citation for the preceding image starts]: Facial angiofibromasCourtesy of Dr Francis J. DiMario Jr [Citation ends].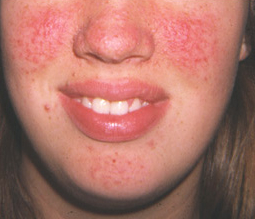
cephalic plaque(s)
Cephalic plaques are hamartomatous patches of vascular and connective tissue with collagen deposition.
These are typically on the forehead or scalp line, and are usually present at birth and enlarge and calcify by adulthood.[Figure caption and citation for the preceding image starts]: Forehead plaqueCourtesy of Dr Francis J. DiMario Jr [Citation ends].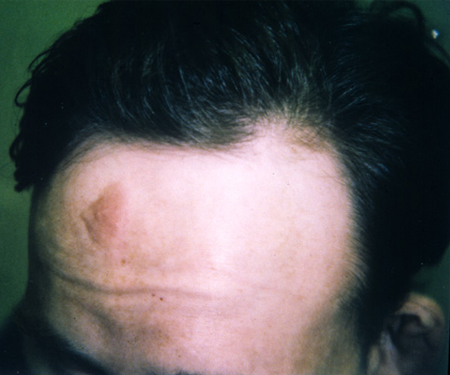
non-traumatic ungual or periungual fibromas
These connective tissue hamartomas can be multiple or isolated.
They are found on the toes more than the fingers, and on females more than males.
They usually develop in the second decade of life.
They are noted in about 20% of patients with TSC.[34]
May be trauma-induced in patients without TSC.[2][3][33][Figure caption and citation for the preceding image starts]: Ungual fibromaCourtesy of Dr Francis J. DiMario Jr [Citation ends].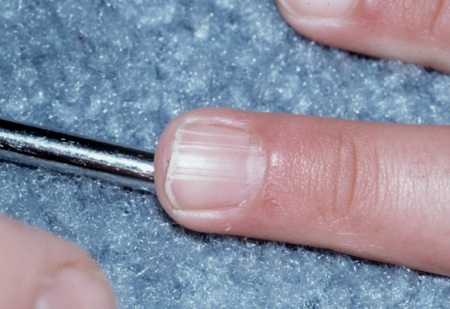
hypomelanotic macules
Three spots are seen in about 90% of patients.[35] The classic ash leaf spot takes on the characteristic pyramidal shape, with a rounded bottom and pointed end. These can generally be seen unaided, but visualisation can be enhanced with the use of a Wood’s lamp.[Figure caption and citation for the preceding image starts]: Hypomelanotic macules and shagreen patchCourtesy of Dr Francis J. DiMario Jr [Citation ends].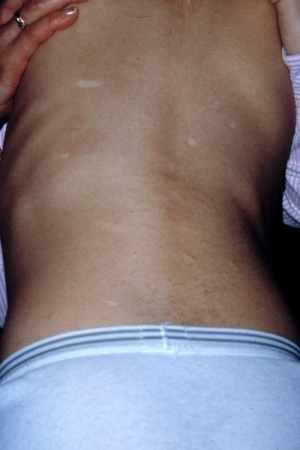
Studies of the skin areas have identified abnormal sudomotor function, with decreased sweat volume and abnormal postganglionic sympathetic innervation.[2][3][33]
shagreen patch(es) (connective tissue nevus)
Most often located in the lumbosacral flank, but may also be found elsewhere.[Figure caption and citation for the preceding image starts]: Hypomelanotic macules and shagreen patchCourtesy of Dr Francis J. DiMario Jr [Citation ends].
Usually evident by 10 years of age and can be dominantly inherited without TSC.
retinal nodular hamartoma(s)
Identified through dilated funduscopic ophthalmological examination.
Composed of glial astrocytic fibres or an achromic patch, these usually cause no visual disturbance and are evident by 2 years of age. They may evolve from being translucent to having calcification.
The lesions are noted in about 50% to 65% of patients with TSC, and are bilateral in 50% of patients.[2][3][33]
[Figure caption and citation for the preceding image starts]: Retinal hamartomaCourtesy of Dr Francis J. DiMario Jr [Citation ends].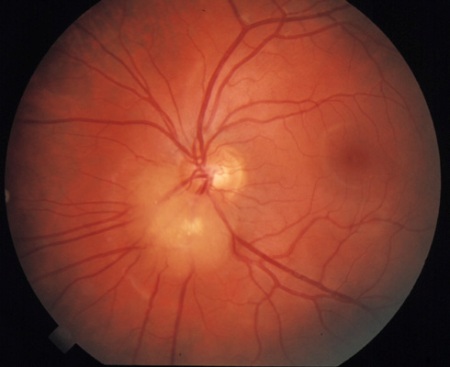
uncommon
polycystic kidney disease
Polycystic kidney disease is infrequently encountered in patients with TSC (approximately <5%).[27][Figure caption and citation for the preceding image starts]: Polycystic kidneysCourtesy of Dr Francis J. DiMario Jr [Citation ends].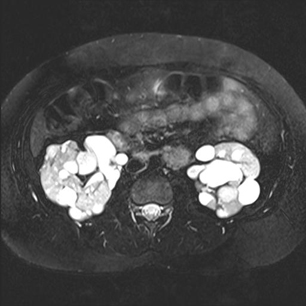 The likelihood is greatest for patients with TSC2, because it results from a contiguous gene deletion/mutation of the PKD1 gene that is immediately adjacent to the TSC2 gene.
The likelihood is greatest for patients with TSC2, because it results from a contiguous gene deletion/mutation of the PKD1 gene that is immediately adjacent to the TSC2 gene.
It is often suspected by the palpation of enlarged kidneys in the newborn, and is confirmed with renal imaging.
Complications during early life include hypertension, flank pain, renal failure, and, less frequently, haematuria.
Because people with polycystic kidneys have ever-increasing enlargement of the cysts, the functional renal parenchyma becomes progressively thinned and dysfunctional until there is renal failure and a need for transplantation.
Preservation of renal function, by minimising the risk for urinary tract infections and the acquisition of renal stones, is paramount.[2][3][26]
Other diagnostic factors
common
numerous dental enamel pits and intraoral fibromas
Virtually all patients with TSC have dental pitting in permanent teeth.[36] These pits are often relatively large holes and multiple in number over the enamel surface.
They do not predispose to excess caries, but may be helpful as an aid to screen asymptomatic first-degree relatives of TSC patients.
Isolated pits can be seen in unaffected people, however, and thus are not specific for TSC.
Intraoral fibromas can be noted in up to 70% of adults with TSC.[36] If these are large, local bleeding and disruption of normal tooth alignment can occur. Pits and intraoral fibromas together are unusual in patients without TSC.[2]
autism
Autism spectrum disorder is seen in about 40% to 50% of patients.[6]
In patients with both TSC and autistic spectrum disorders, there is a 75% prevalence of cognitive impairment and a 75% to 100% prevalence of concurrent epilepsy. In combination this group of clinical problems is referred to as the TSC-associated neuropsychiatric disorders (TAND).[2][3][7][38]
cognitive impairment
uncommon
multiple hamartomatous colonic polyps
These polyps are uncommonly identified, but may be present in many patients with TSC. A true prevalence is not known.
A biopsy is required to confirm the hamartomatous histology, which should distinguish them from adenomatous polyps seen in other diseases.[37]
Risk factors
strong
genetic predisposition
Approximately one-third of people with TSC have autosomal-dominant inheritance, while the remaining two-thirds of cases are due to sporadic mutations.[4]
Use of this content is subject to our disclaimer