Investigations
1st investigations to order
brain MRI
Test
MRI should be ordered as soon as a rapidly progressive dementia is suspected and should include T1, T2, diffusion-weighted imaging (DWI), attenuated diffusion coefficient map (ADC), and fluid-attenuated inversion recovery (FLAIR) sequences.[76] If possible, DWI and ADC sequences should be acquired in both coronal and axial planes to minimise air-brain interface artifact. [Figure caption and citation for the preceding image starts]: Changes in the basal ganglia seen in Creutzfeldt-Jakob disease. (A) Fluid-attenuated inversion recovery MRI and (B) diffusion-weighted MRI of the same patient demonstrate bilateral basal ganglia hyperintensities (arrows). There is also mild bilateral medial thalamus and pulvinar hyperintensityFrom the personal collection of Dr M. Geschwind [Citation ends].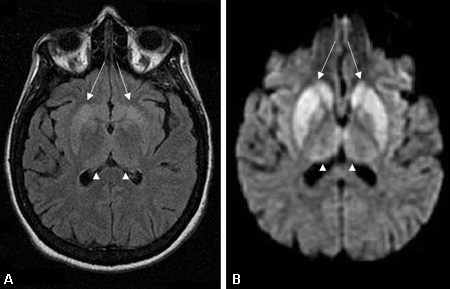 [Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].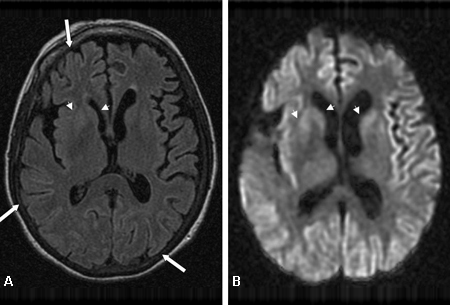
Abnormalities found on the ADC and DWI MRI sequences in sporadic Creutzfeldt-Jakob disease (sCJD) and variant CJD (vCJD) have generally not been reported in other similar dementias and are strongly suggestive of CJD.[67]
If the scan is of good quality, MRI, DWI, and FLAIR scans can have about 92% to 96% sensitivity and 93% to 94% specificity.[65][67][77]
On T1 images with gadolinium, CJD generally does not show contrast enhancement, or white-matter intensity abnormalities; if these are found, other diagnoses should be investigated.[78][79]
T1 hypointensity has been reported in the globus pallidus in CJD.[87] The pulvinar sign, a term referencing bilateral pulvinar hyperintensity, can be seen in vCJD. When both the pulvinar and medial thalamic show signal intensity, vCJD should be suspected.[71][80][81] Certain forms of familial CJD and other genetic prion diseases (particular mutations) have similar MRI findings to those seen on DWI MRI in sCJD, but many genetic prion diseases just show atrophy.
Result
often demonstrates hyperintensity in the cerebral cortex (cortical ribboning), basal ganglia (caudate and putamen), and thalamus on diffusion-weighted imaging (DWI) and fluid-attenuated inversion recovery (FLAIR) sequences, and hypointensity (restricted diffusion) on attenuated diffusion coefficient map (ADC) sequences
EEG
Test
Abnormalities, although moderately specific, are only about 60% sensitive and may not appear until the later stages of the disease.[14][82]
When other conditions with similar EEG abnormalities have been ruled out, these findings can have high specificity for prion disease.[71]
Result
generalised slowing, focal or diffuse, and periodic sharp-wave complexes
Investigations to consider
quaking-induced conversion (QuIC)
Test
Sensitive and specific test that detects the disease-associated isoform of the prion protein in CSF from patients with sporadic CJD.[68] Real time QuIC (RT-QuIC) and end-point QuIC (EP-QuIC) are the versions currently used by diagnostic laboratories, depending on country.
Result
positive
CSF biomarkers
Test
Although helpful in confirming rapid neuronal deterioration, the CSF biomarkers cannot definitively diagnose or rule out prion disease.[83][88][89][90] These tests should be interpreted with caution as their sensitivity and specificity for sCJD are still unclear.
Abeta-42 protein may be diminished and total tau (T-tau) and phosphorylated tau (P-tau) may be elevated in Alzheimer's disease.
The 14-3-3 protein found in CSF has been reported as being a strong indicator of CJD. However, the sensitivity and specificity of this test varies greatly in the literature.[71][91][92][93] Current guidelines are shifting away from the use of this test.
Some studies suggest that the CSF proteins, such as 14-3-3 protein, total tau, and neuron-specific enolase, have higher sensitivity in rapidly progressive diseases, supporting the idea that they are released during neuronal death and injury and are not necessarily specific for prion disease.[71][84][88][89]
Result
14-3-3 protein (i.e., western blot) may be positive; total tau (T-tau) protein elevated ( >1200 picograms/mL); neuron-specific enolase elevated (>35 nanograms/mL)
prion protein gene genetic testing
Test
Onset and clinical manifestations of all forms of prion disease are strongly influenced by the polymorphic codon 129 of the endogenous prion protein gene (PRNP). Codon 129 can be either methionine (M) or valine (V). Homozygotic combinations (e.g., MM or VV) result in higher risk for developing prion disease. The prion type (type 1 or 2) also influences the disease presentation; however, the prion typing can only be determined from frozen brain tissue obtained by either brain biopsy or autopsy.[14][23]
Genetic prion diseases are caused by a mutation in the gene encoding PRNP, located on chromosome 20.[6] To date more than 40 different PRNP mutations have been identified, each presenting with its own disease phenotype (i.e., Gerstmann-Straussler-Scheinker, fatal familial insomnia, and familial CJD).
PRNP mutations are transmitted in an autosomal dominant manner.[6] Importantly, about 60% of patients with prion disease that is found to be genetic were not known to have a positive family history of prion disease. Further inspection will often find a family history of Alzheimer's or Parkinson's disease that was likely misdiagnosed or a parent who died before the onset of symptoms.[21][22]
Patients and families also need to undergo genetic counselling and understand the implications involved before learning such results. As genetic prion disease is autosomal dominant, the Huntington's disease protocol is generally followed.[86] This protocol is used for many autosomal dominant neurological disorders to make sure that the patients, their families, and others understand the psychological, psychiatric, medical, legal, and other implications of genetic testing.
Samples should be accompanied by a brief clinical history and shipped the same day of collection.
The National Creutzfeldt-Jakob Disease Research and Surveillance Unit Opens in new window Public Health Agency of Canada: prion disease information Opens in new window National Prion Disease Pathology Surveillance Center Opens in new window
Result
positive
biopsy (brain, tonsil)
Test
Brain biopsy is the only definitive way to diagnose sporadic prion disease antemortem. In variant CJD, a tonsil biopsy can be diagnostic.
Due to the unpredictable pattern of protein accumulation in the brain, a false-negative result is possible.
Brain biopsy procedures may place patients at unnecessary risk for infection or further brain damage. If the diagnosis is found to be true, there is still no available treatment.
Prion proteins are resistant to standard surgical sterilisation methods, and medical staff performing the procedure may be placed at risk for CJD transmission. Appropriate protocols to minimise transmission should be followed.[27]
All things considered, it is not recommended that brain biopsy be done to diagnose CJD, especially as the clinical history, MRI and QuIC are now so helpful with diagnosis.[67][71]
If brain biopsy is done, tissue should be taken from areas of MRI abnormality where possible.
Although the microscopic hallmarks are characteristic of this disease, other conditions can also have these microscopic changes.
The presence of PrPSc by immunochemistry or western blot is required for definitive diagnosis.[3][36]
In approximately 10% of CJD, amyloid plaques of PrPSc are found, and in cases of Gerstmann-Straussler-Scheinker an amyloid PrPSc core is surrounded by another group of smaller amyloid globules.
Variant CJD (vCJD) also has a relatively unique pathological feature of core PrPSc amyloid plaques surrounded by vacuoles. These are called florid plaques, as they resemble a flower.
In vCJD, PrPSc can also be identified in the lymphoreticular system during the disease course, and tonsil biopsies may show the presence of PrPSc in vCJD, although not other forms of human prion disease.[3][37]
Result
microscopic: vacuolation (spongiform changes), neuronal loss, and astrogliosis; histological: presence of pathogenic prion (PrPSc) by immunohistochemistry or western blot; may show amyloid
Use of this content is subject to our disclaimer