Differentials
Alzheimer's dementia (AD)
SIGNS / SYMPTOMS
Generally a more chronic and rarely rapidly progressing dementia, but atypical cases can be mistaken for Creutzfeldt-Jakob disease (CJD).[94]
CJD patients commonly present after 60 years of age, with deficits in memory and cognition in the absence of other disorders.
Clinical features of CJD that are not commonly seen in AD include subacute onset and/or difficulty in co-ordination early on in the course.[95]
INVESTIGATIONS
AD is a clinical diagnosis. The clinical history, performance on neuropsychological testing, and pattern of brain MRI atrophy, single photon emission computed tomography (SPECT) hypoperfusion, or PET scan hypometabolism may support the diagnosis of AD. An MRI that is diagnostic for CJD may rule out AD. [Figure caption and citation for the preceding image starts]: Changes in the basal ganglia seen in Creutzfeldt-Jakob disease. (A) Fluid-attenuated inversion recovery MRI and (B) diffusion-weighted MRI of the same patient demonstrate bilateral basal ganglia hyperintensities (arrows). There is also mild bilateral medial thalamus and pulvinar hyperintensityFrom the personal collection of Dr M. Geschwind [Citation ends].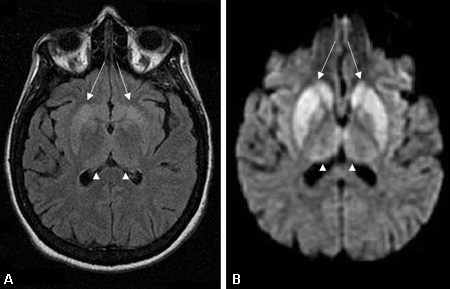 [Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends]. [Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].
[Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].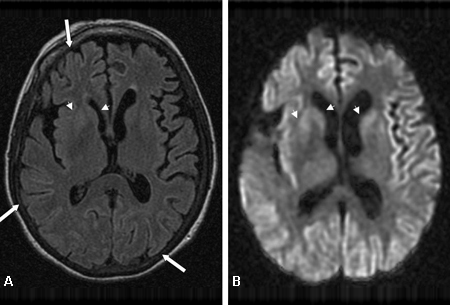
PET scanning with Pittsburgh compound B (may soon be a diagnostic test for AD.[96]
Lewy body dementia
SIGNS / SYMPTOMS
In a research study with a large cohort, dementia with Lewy bodies was found to be the second most commonly mistaken dementia for Creutzfeldt-Jakob disease (CJD).[84][97]
Like CJD, dementia with Lewy bodies is a neurodegenerative disease that can present with cognitive impairment, visual hallucinations or disturbances, and parkinsonism.[98][99]
INVESTIGATIONS
This condition is best distinguished from CJD by diffusion-weighted imaging (DWI), attenuated diffusion coefficient map (ADC), or fluid-attenuated inversion recovery (FLAIR) imaging on brain MRI.[Figure caption and citation for the preceding image starts]: Changes in the basal ganglia seen in Creutzfeldt-Jakob disease. (A) Fluid-attenuated inversion recovery MRI and (B) diffusion-weighted MRI of the same patient demonstrate bilateral basal ganglia hyperintensities (arrows). There is also mild bilateral medial thalamus and pulvinar hyperintensityFrom the personal collection of Dr M. Geschwind [Citation ends].[Figure caption and citation for the preceding image starts]: Bilateral medial thalamus and pulvinar hyperintensity (arrowheads) on (A) fluid-attenuated inversion recovery and (B) diffusion-weighted MRI in a patient with Creutzfeldt-Jakob. This patient also has significant basal ganglia hyperintensity on both sequences (arrows)From the personal collection of Dr M. Geschwind [Citation ends].[Figure caption and citation for the preceding image starts]: Diffuse cortical ribboning (arrows) seen on (A) diffusion-weighted imaging (DWI) and less so on (B) fluid-attenuated inversion recovery (FLAIR) MRI. Both sequences show cerebral cortex gyral hyperintensitiesFrom the personal collection of Dr M. Geschwind [Citation ends].
SPECT or PET scan may also reveal low dopamine transporter uptake in the basal ganglia.[99]
Periodic sharp wave complexes are rarely seen on EEG in the late stages of Lewy body dementia.
Frontotemporal dementia
SIGNS / SYMPTOMS
Although frontotemporal dementia (FTD) typically has a faster course than most memory and ageing disorders, it is seldom as rapidly progressive as Creutzfeldt-Jakob disease (CJD).
Patients usually present with a frontal syndrome, including behavioural, personality, and cognitive changes, before the onset of dementia.[100][101][102]
Unlike CJD, criteria for frontotemporal dementia exclude myoclonus and cerebellar ataxia.[103]
INVESTIGATIONS
FTD is a clinical diagnosis. The clinical history, neurological examination, performance on neuropsychological testing, and pattern of brain MRI atrophy, SPECT hypoperfusion, or PET scan hypometabolism may support a diagnosis of FTD.
An MRI (DWI and ADC sequences) that is diagnostic for CJD essentially rules out FTD.
Corticobasal syndrome (CBS)
SIGNS / SYMPTOMS
Another progressive dementia that typically has a slower course than Creutzfeldt-Jakob disease (CJD).
Visual, sensory, and motor deficits of corticobasal degeneration (CBD) may initially suggest CJD.[71][104]
CBS is typically due to an underlying pathology of CBD or Alzheimer's disease (AD), and rarely CJD.
INVESTIGATIONS
CBS is a clinical diagnosis, usually with an underlying pathological diagnosis of CBD or AD. The clinical history, neurological examination, performance on neuropsychological testing, and pattern of brain MRI atrophy, SPECT hypoperfusion, or PET scan hypometabolism may support a diagnosis of CBS.
An MRI that is diagnostic for CJD rules out CBS.
Vasculitis
SIGNS / SYMPTOMS
Vasculitides can cause dementia or encephalopathy when they affect the CNS. These disorders can be distinguished from Creutzfeldt-Jakob disease (CJD) by the presence of systemic or peripheral nervous system signs such as fever, weight loss, neuropathy, and organ involvement.
Criteria used in the diagnosis of such disorders have been established by the American College of Rheumatology.[71][105][106][107]
INVESTIGATIONS
ESR and CRP may be elevated.
RF and ANA may be positive.
Perinuclear antineutrophilic cytoplasmic antibody (P-ANCA) and cytoplasmic antineutrophilic cytoplasmic antibody (C-ANCA) may be positive.
Brain angiogram demonstrates an intermittent narrowing 'sausage' appearance not seen in CJD.
CSF may show evidence of inflammation, such as pleocytosis.
Meningeal and brain biopsy shows inflammation of the blood vessels with lymphocytic infiltrate that is not present in CJD.
Although serological rheumatological evaluation should be performed for elevated auto-antibodies, in isolated CNS vasculitis, these blood tests may be negative and systemic signs may be absent.[108]
Paraneoplastic limbic encephalitis and non-paraneoplastic autoimmune limbic encephalitis
SIGNS / SYMPTOMS
Autoimmune disorders that mimic prion diseases include paraneoplastic limbic encephalitis and non-paraneoplastic autoimmune limbic encephalitis.[109]
Paraneoplastic limbic encephalopathies typically occur in 2 forms: antibody-mediated autoimmune conditions or non-antibody autoimmune-mediated causes such as systemic (but non-CNS) cancers.
Paraneoplastic disorders may present in patients with known or family history of cancer, or even precede the detection of cancer entirely.
If a paraneoplastic condition is suspected, body CT imaging with contrast and testicular ultrasound (where appropriate) is indicated .[71][110][111][112]
About 30% of these patients also present with seizures.[109][110][111]
INVESTIGATIONS
Serum and CSF should be tested for elevated tumour markers and the presence of paraneoplastic antibodies.
ESR and CRP may be elevated.
RF and ANA may be positive.
P-ANCA and C-ANCA may be positive.
Tumour markers such as CEA, CA-125, or PSA may be elevated.
Paraneoplastic antibodies present: anti-Hu (ANNA-1), anti-Ta (anti-Ma2), anti-CV2 (anti-CRMP-5), anti-amphiphysin, anti-Yo (PCA-1), anti-nCMAg, anti-Ma1, anti-Ri (ANNA-2), anti-AMPAR, anti-N-methyl-D-aspartate receptor (NMDAR), anti-voltage-gated potassium channel (VGKC)-associated antibodies.
Limbic encephalopathy autoimmune antibody-mediated causes not related to cancers
SIGNS / SYMPTOMS
Limbic encephalopathy can also be due to autoimmune antibody-mediated causes not associated with cancers, as seen in anti-VGKC antibodies and other antineutrophil antibodies, and Hashimoto's encephalopathy.
Sometimes these antibodies are associated with cancers (paraneoplastic) and sometimes they are not (non-paraneoplastic).[109][112][113]
INVESTIGATIONS
VGKC and anti-glutamic acid decarboxylase (GAD) antibodies are elevated.
Antithyroid peroxidase (ATPO) or anti-thyroglobulin (ATG) antibodies are markedly elevated.
Hashimoto's encephalopathy
SIGNS / SYMPTOMS
Rare but treatable autoimmune disorder that is commonly mistaken for Creutzfeldt-Jakob disease (CJD). Linked to chronic lymphocytic thyroiditis, this disease should be suspected with the presence of elevated antithyroid auto-antibodies.
Although this disorder is also a rapidly progressive dementia with many common features of CJD, it is distinguished by its fluctuating course and more common association with seizures.[109][114][115]
Patients may be euthyroid, subclinically hypothyroid, hypothyroid, or hyperthyroid, but to make the diagnosis, patients must be treated so that they are in a euthyroid state.[113]
Hashimoto's encephalopathy is a diagnosis of exclusion.
As the antithyroid antibodies are not shown to be causative in Hashimoto's encephalopathy, other terms have been applied to this condition, such as non-vasculitic autoimmune inflammatory meningoencephalitis and corticosteroid-responsive encephalopathy associated with autoimmune thyroiditis.
These autoimmune-mediated disorders are very important to identify, as they are readily treatable with immunosuppression, such as with high-dose corticosteroids.[109][113][116][117][118][119]
INVESTIGATIONS
Thyroid dysfunction may be present.
Antithyroid peroxidase (ATPO) and/or anti-thyroglobulin (ATG) antibodies are positive.
Brain MRI (ADC and DWI sequences) does not show the abnormalities seen in CJD.
Acid-base and electrolyte disorders
SIGNS / SYMPTOMS
When patients present with recent-onset dementia of unknown aetiology, a toxic-metabolic encephalopathy work-up should be done.
Metabolic disturbances such as electrolyte imbalances can produce rapidly progressive dementia.
INVESTIGATIONS
Potassium, sodium, calcium, or magnesium levels are abnormal.
Other metabolic conditions
SIGNS / SYMPTOMS
Other metabolic conditions in the Creutzfeldt-Jakob disease (CJD) differential include childhood metabolic disorders that may present in adults as dementia such as porphyria, adult-onset metachromatic leukodystrophy, orthochromatic leukodystrophies, and Kufs' disease.
Systemic symptoms associated with these conditions are usually slower progressing but may be accompanied by rapid cognitive decline.[120]
INVESTIGATIONS
Urine porphobilinogens are elevated during episodes of porphyria.
Skin biopsy with electron microscopy is recommended for Kufs' disease diagnosis.
Urine arylsulfatase levels are depressed in metachromatic leukodystrophy.
Serum long-chain fatty acids are elevated in adrenal leukodystrophy.
Vitamin B1 deficiency
SIGNS / SYMPTOMS
When patients present with recent-onset dementia of unknown aetiology, a toxic-metabolic encephalopathy work-up should be done.
Metabolic disturbances such as certain vitamin deficiencies or rapid sodium shifts (causing extrapontine myelinolysis) can produce rapidly progressive dementia.
A thiamine (vitamin B1) deficiency should be urgently considered in patients who are nutritionally deprived with neurological signs.
Inadequate thiamine in the nervous system can lead to Wernicke's encephalopathy, which presents with nystagmus, ataxia, and memory loss.
INVESTIGATIONS
Erythrocyte thiamine level is low.
Brain MRI (T2-weighted, FLAIR, DWI, and ADC) may show abnormalities (restricted diffusion) in the mammillary bodies, thalami, periaqueductal white matter, and/or tectum. MRI findings in vitamin B1 deficiency have some overlap with those in sporadic Creutzfeldt-Jakob disease (sCJD), including restricted diffusion in the thalamus and other deep nuclei.
Vitamin B3 deficiency
SIGNS / SYMPTOMS
Deficiencies of niacin or vitamin B3 (or the amino acid tryptophan, which is converted into niacin), may result in pellagra, which may present as a triad of dermatitis, diarrhoea, and dementia.
Although the onset of pellagra is more insidious (death within a few years), it should be considered in nutritionally compromised (particularly low-protein intake) patients with dementia.
INVESTIGATIONS
Nicotinic acid metabolites in the urine can confirm diagnosis; patients are treated with niacin supplements.
Vitamin B12 deficiency
SIGNS / SYMPTOMS
In general, all patients with dementia should be tested for vitamin B12 levels, as this condition can respond to treatment.[121]
INVESTIGATIONS
Low serum vitamin B12.
The hallmark of diagnosis is the presence of a macrocytic anaemia with macro-ovalocytes in the peripheral blood smear.
Vitamin E deficiency
SIGNS / SYMPTOMS
Vitamin E deficiencies can occur in people who are unable to absorb or metabolise fat-soluble vitamins.
In rare cases, it is caused by an autosomal recessive mutation in the alpha-tocopherol transfer protein gene. This deficiency can cause ataxia and other movement disorders such as dystonia. It is rarely rapidly progressive and often presents similarly to Friedreich's ataxia.
When it is diagnosed early, treatment with vitamin E can prevent progression.[122]
INVESTIGATIONS
Serum vitamin E level is low.
Wilson's disease
SIGNS / SYMPTOMS
Wilson's disease is due to an autosomal recessive mutation that hinders copper metabolism.
The accumulation of copper in the tissues eventually causes dementia and liver disease, and generally presents in the teenage years, but almost always in patients <50 years of age.
Although not rapidly progressive, it is important to consider this in younger adults with cognitive, behavioural, and/or movement disorder, as it is very treatable.[123][124]
INVESTIGATIONS
Serum and urine copper elevated.
Blood ceruloplasmin is low.
Heavy metal intoxication
SIGNS / SYMPTOMS
Heavy metal intoxication, especially with acute exposure, can lead to rapid cognitive decline.
In contrast to rapidly progressive dementias, which progress over weeks to months, these encephalopathies can progress within hours to days.
Patients being treated with bismuth (a metal used to treat GI disorders) should also be tested for toxicity. Often mistaken for Creutzfeldt-Jakob disease (CJD), bismuth intoxication can cause ataxia, apathy, and eventually myoclonus, speech problems, and change in mental status. If not treated, this condition can lead to permanent tremors and/or death.[125][126][127][128]
INVESTIGATIONS
Patients should be screened for arsenic, mercury, aluminium, lithium, bismuth, and lead toxicities.
Hepatic encephalopathy
SIGNS / SYMPTOMS
Hepatic encephalopathy should be clinically easy to distinguish from Creutzfeldt-Jakob disease (CJD) because of presence of severe underlying liver disease.
INVESTIGATIONS
Serum lactate and ammonia elevated.
The EEG in hepatic encephalopathy may show periodic sharp waves similar to CJD.
HIV-related mental status changes
SIGNS / SYMPTOMS
One fourth of AIDS patients eventually develop a neurological condition such as AIDS-dementia complex, HIV encephalopathy, or HIV-associated dementia. Therefore, all patients with rapidly progressive dementias should consider HIV testing.[71]
Patients generally do not present with the combination of cognitive, motor, vision, and behavioural abnormalities as seen in Creutzfeldt-Jakob disease (CJD), but there is some overlap.
INVESTIGATIONS
HIV 1 and 2 antibody screen is positive.
Brain MRI does not show features consistent with CJD on diffusion imaging.
Syphilis
SIGNS / SYMPTOMS
Spirochete infections are an unusual but treatable cause of dementia. Patients should be tested for Treponema pallidum or neurosyphilis, as cognitive dysfunction is a late complication of syphilis.[129]
Patients generally do not present with the combination of cognitive, motor, vision, and behavioural abnormalities as seen in Creutzfeldt-Jakob disease (CJD), but there is some overlap. Also, they generally do not have rapidly progressive dementia.
INVESTIGATIONS
CSF VDRL test is reactive.
FTA-ABS test is reactive.
Brain MRI does not show features consistent with CJD on diffusion imaging.
Lyme disease
SIGNS / SYMPTOMS
Lyme disease is an infection caused by a tick bite containing the spirochete Borrelia burgdorferi. Neurological and psychiatric manifestations in this systemic infection occur with neurological involvement.[130]
Although rarely reported as a rapidly progressive dementia, it should still be considered, as it is readily treatable.[131][132][133]
Typically presents as a skin lesion indistinguishable from erythema migrans, and is principally found in the southeast and south central regions of the US.
INVESTIGATIONS
B burgdorferi antibody elevated above index positive values.
Whipple's disease
SIGNS / SYMPTOMS
Whipple disease, a rare bacterial infection caused by Tropheryma whipplei, interferes with many body systems and can manifest as a neuropsychiatric syndrome.
Typically occurring at around 50 years of age, this condition predominantly affects men and can vary in its clinical presentation. Most commonly, patients present with gastrointestinal disturbances, cognitive impairment, and problems walking.
May be mistaken for progressive supranuclear palsy due to the behavioural and eye movement abnormalities.[134]
Once Whipple disease is diagnosed, patients can be treated with antibiotics.[135][136]
INVESTIGATIONS
Jejunal biopsy indicates presence of foamy macrophages.
Tropheryma whipplei PCR of CSF positive.
CNS malignancy
SIGNS / SYMPTOMS
Several malignancies can cause rapidly progressive dementias.
Primary cancers that can mimic Creutzfeldt-Jakob disease (CJD) include primary CNS lymphoma, intravascular lymphoma, and gliomatosis cerebri.
These conditions may present with many features of CJD and may be clinically indistinguishable.
INVESTIGATIONS
CSF cytology and flow cytometry demonstrate abnormal cells (CNS lymphoma).
Patients should be screened with brain MRI (with and without contrast); CT scans of the chest, abdomen, and pelvis with contrast; whole-body PET scan; mammogram; and CSF and serum cancer screens.
Obvious brain masses should be easily distinguishable from CJD.[121]
Psychiatric disorders
SIGNS / SYMPTOMS
Many psychiatric disorders can present in adulthood or later in life, but do not have accompanying neurological abnormalities.
Many patients who self-refer themselves to physicians because they think they have Creutzfeldt-Jakob disease or another prion disease have anxiety or conversion disorders.
INVESTIGATIONS
A thorough neurological examination to identify any true neurological abnormality should generally exclude a psychiatric disorder.
Brain MRI showing abnormalities would generally not be consistent with psychiatric disease.
Progressive supranuclear palsy
SIGNS / SYMPTOMS
When evaluating patients with rapidly progressive symptoms, consider Progressive Supranuclear Palsy (PSP) if there is early onset of unprovoked falls, particularly backward, typically within the first 1-3 years of symptom onset. PSP is also characterised by vertical supranuclear gaze palsy and slow vertical saccades, causing difficulty with downward gaze.
Symmetric parkinsonism with predominant axial rigidity and poor or transient response to levodopa is typical for PSP.
Additionally, patients may exhibit dysphagia, dysarthria, and cognitive impairment mainly involving frontal executive dysfunction, such as difficulty with planning and multitasking.
While both prion diseases and PSP show rapid progression, PSP generally progresses over several years, whereas prion diseases can progress much more rapidly, often within months.
Speech/language disorders, including non-fluent/agrammatic primary progressive aphasia or progressive apraxia of speech, and frontal behavioural changes, such as apathy, disinhibition, or perseveration, may also be evident early in PSP.[137][138]
INVESTIGATIONS
Midbrain atrophy is significant on MRI, displaying characteristic radiographical signs such as the 'hummingbird' and 'Mickey Mouse' signs in the midsagittal and axial planes. These findings are frequent in PSP but not typical in prion diseases.
Definitive diagnosis of PSP requires postmortem histopathological evidence, showing tauopathy with neurofibrillary tangles and tufted astrocytes. In contrast, prion diseases show spongiform changes and prion protein deposition.
Vertical saccade velocity assessment can identify the slow eye movements characteristic of PSP, which are not typical in prion diseases.
A poor or transient response to high doses of levodopa is typical for PSP and is not usually seen in prion diseases.
Supportive imaging findings in PSP include postsynaptic striatal dopaminergic degeneration observed on PET or SPECT imaging, which are not characteristics of prion diseases.
If disease progression is particularly rapid, CSF biomarkers to exclude prion disease may be indicated. RT-QuIC has the highest power to discriminate these two conditions, but a very high total tau is also suggestive of prion disease instead of PSP, where tau levels are usually decreased.[137]
Use of this content is subject to our disclaimer