Aetiology
The aetiology of neuroblastoma remains unclear. No specific environmental exposure has been implicated in the development of neuroblastoma.
Approximately 1% to 2% of patients have a family history of neuroblastoma.[13] Most hereditary neuroblastomas are caused by alterations in the ALK (anaplastic lymphoma kinase) gene.[14] Germline PHOX2B (paired-like homeobox 2b) mutations are a relatively rare cause of hereditary neuroblastoma.[15][16]
A few medical conditions, some of which are related to aberrant neural crest development, predispose a patient to developing neuroblastoma, including Turner syndrome, Hirschsprung's disease, congenital central hypoventilation syndrome, and neurofibromatosis type 1.[17][18]
Pathophysiology
Neuroblastoma is classically an embryological neural crest-derived malignancy. The neural crest is a group of neuronal cells that migrate from the spinal cord to form many structures, including the sympathetic nervous system, during fetal development. Therefore, the tumour is found to originate from sympathetic ganglia near the spinal cord and within the adrenal medulla. As the glycolipid disialoganglioside (GD2) is expressed during differentiation into sympathetic nervous tissue, GD2 is typically found on the surface of neuroblastomas. GD2 is a therapeutic target.[19][20]
The majority of tumours arise in the abdomen, most commonly in the adrenal gland(s). However, tumours may also present with thoracic or paravertebral primary sites, from the neck to the pelvis.[21] Very rarely, a primary site cannot be identified. Neuroblastoma can metastasise by both lymphatic and haematological spread, the most common sites being lymph nodes, bone marrow, bone, liver, skin, orbits, and dura. Intracranial and pulmonary spread is also possible, but infrequent.[22]
Neuroblastoma is highly heterogeneous, due in part to the fact that it arises from a tissue type that is undergoing rapid differentiation during fetal development, and the transition from normal to malignant tissue can occur at multiple points in development. Therefore, some tumours are rapidly proliferative but regress over time and other tumours grow more slowly but are highly malignant.[23]
Several oncogenes involved in neural crest development are implicated in the development of neuroblastoma (e.g., v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog [MYCN], anaplastic lymphoma kinase [ALK], and paired-like homeobox 2b [PHOX2B]).[14][15][16][24] These genes are also involved in neural crest development.[23][Figure caption and citation for the preceding image starts]: Neural crest development and neuroblastoma formation; EMT = epithelial to mesenchymal transitionCreated by BMJ Knowledge Centre, based on Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med 2015;357:49-63; used with permission [Citation ends].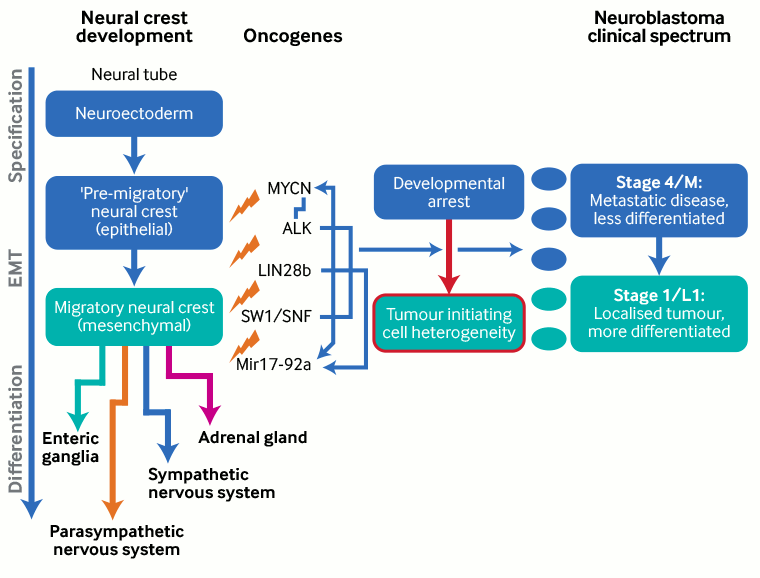
Most neuroblastoma tumours retain the ability to metabolise catecholamines (e.g., adrenaline/epinephrine, noradrenaline/norepinephrine, and dopamine), which can lead to hypertension or other symptoms associated with catecholamine excess (e.g., dizziness, nausea, headache).[25] Importantly, the metabolites homovanillic acid (HVA) and vanillylmandelic acid (VMA) are secreted by the majority of neuroblastoma tumours and can be detected in patient's urine.[26]
Classification
International Neuroblastoma Pathology Classification[4]
Degree of differentiation:
Neuroblastic tumours are classified as follows:
Neuroblastoma
Ganglioneuroblastoma (nodular or intermixed)
Ganglioneuroma.
These tumour types represent a spectrum of neural differentiation from primitive neuroblasts: neuroblastoma (the most common type); a mixture of immature neuroblasts and ganglion cells, as seen in ganglioneuroblastoma; and the mature ganglion cells that are present in ganglioneuroma.
Degree of differentiation is a component of risk stratification and increased differentiation has been associated with a better prognosis.
Histology:
Tumours are classified as either having favourable or unfavourable histology:
Favourable histology: includes the presence of more differentiation towards ganglion cells or poorly differentiated tumours with low or intermediate mitosis-karyorrhexis index (MKI) and young patient age (<1.5 years).
Unfavourable histology: includes undifferentiated tumours, high MKI, older patient age, nodular ganglioneuroblastoma, and poorly differentiated tumours with low or intermediate MKI and older patient age (≥1.5 years).
Use of this content is subject to our disclaimer