Approach
The diagnosis of primary myelofibrosis (PMF) is one of exclusion because of the lack of a specific diagnostic marker. Once a diagnosis of PMF is made, it is important to carry out risk assessment to inform prognosis and to guide management.
History and physical examination
A careful history and physical examination should be performed to identify signs and symptoms associated with PMF, and to identify a reactive cause or another disorder that may suggest secondary myelofibrosis (e.g., polycythaemia vera [PV], essential thrombocythaemia [ET], systemic mastocytosis, acute leukaemia, hairy cell leukaemia, hyperparathyroidism, systemic lupus, drug treatment, or toxic agent).
Ruling out secondary myelofibrosis is essential because prognosis and management of other causes/conditions associated with myelofibrosis (e.g., post-PV and post-ET myelofibrosis) will differ.[34]
At diagnosis, PMF patients commonly present with signs and symptoms associated with anaemia (e.g., fatigue, weakness, dyspnoea, palpitations), and constitutional symptoms associated with a hypercatabolic state (e.g., weight loss, night sweats, low-grade fever, cachexia, fatigue, pruritus). Up to 30% of patients may be asymptomatic at diagnosis.[35]
Patients should be examined for splenomegaly, hepatomegaly, extramedullary haematopoiesis, portal hypertension, bleeding, bone and joint changes, and immune dysfunction.
Splenomegaly: a hallmark of PMF and present in virtually every patient with PMF at diagnosis. If absent, other causes of the clinical abnormalities should be considered. The degree of splenomegaly varies but is frequently substantial. Because the rate of splenic enlargement is variable, spleen size cannot be used as an indication of disease duration.[36] Splenomegaly may result in early satiety, generalised abdominal discomfort, and left upper quadrant discomfort. Splenic infarcts, perisplenitis, or subcapsular haematoma may cause severe left upper quadrant or left shoulder pain.
Hepatomegaly: present in 40% to 70% of patients at diagnosis.[37]
Extramedullary haematopoiesis: a hallmark of PMF. Depending on the organ or site of involvement, it results in haemorrhage (gastrointestinal tract haemorrhage, cutaneous petechiae, haemoptysis, haematuria), spinal cord compression, focal seizures, symptoms related to increased intracranial pressure, ascites, pericardial or pleural effusion, pulmonary hypertension, and respiratory failure.
Portal hypertension: can present without signs and symptoms, or manifest as ascites, oesophageal and gastric varices, gastrointestinal bleeding, hepatic encephalopathy, and hepatic or portal vein thrombosis.
Platelet dysfunction, acquired factor V deficiency, thrombocytopenia, and disseminated intravascular coagulation: may occur, contributing to bleeding.
Joint and bone pain, symptoms of osteosclerosis, or gout: may be reported.
Otosclerosis can cause hearing loss.
Infections (most commonly pneumonia): may result from deficiencies in humoral immunity.
Initial investigations
A full blood count (FBC) with differential, and peripheral blood smear, are the first tests to order. Bone marrow aspiration and biopsy are required to establish a diagnosis if history, examination, and initial blood tests suggest PMF.
FBC
An FBC is essential in patients thought to have PMF. Because of its origin in a multipotent haematopoietic progenitor cell, PMF affects all blood cell types but not in a predictable manner.
Anaemia, usually mild, is present in most patients with PMF, with >60% having a haemoglobin concentration <100 g/L (<10 g/dL). A normal haemoglobin or haematocrit in the presence of substantial splenomegaly should lead to immediate consideration of PV because the expanded plasma volume associated with splenomegaly can mask a substantial increase in the red blood cell (RBC) mass.
Leukocyte and platelet counts can be low, normal, or high without reference to spleen size.
The presence of blood count abnormalities should prompt a careful examination of a peripheral blood smear.
Peripheral blood smear
In patients with PMF, peripheral blood smear will usually show immature white cells (metamyelocytes, myelocytes, promyelocytes, myeloblasts), nucleated RBCs, and teardrop-shaped RBCs, as a result of extramedullary haematopoiesis. This leuko-erythroblastic reaction is not specific for PMF, but it is a hallmark of PMF and its absence should challenge the clinical impression.[Figure caption and citation for the preceding image starts]: Peripheral blood smear showing leuko-erythroblastic reaction: teardrop red blood cells (black arrows), myelocyte (red arrow), and promyelocyte (blue arrow)From the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].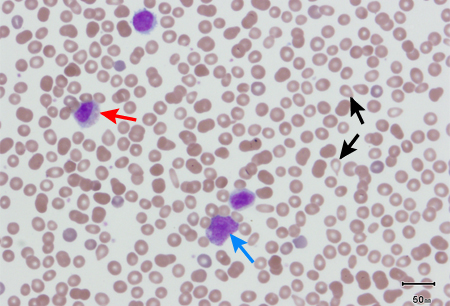 [Figure caption and citation for the preceding image starts]: Peripheral blood smear showing teardrop red blood cells (black arrows), 2 nucleated red blood cells (red arrows), and a myelocyte (blue arrow)From the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Peripheral blood smear showing teardrop red blood cells (black arrows), 2 nucleated red blood cells (red arrows), and a myelocyte (blue arrow)From the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].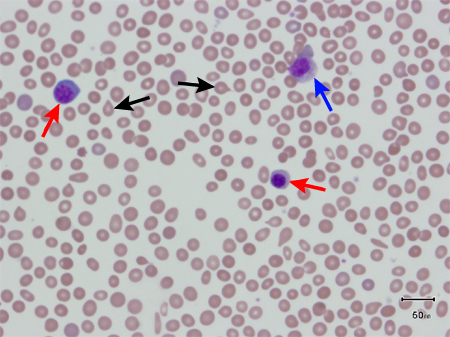
Bone marrow aspiration and biopsy
Essential for establishing a diagnosis. Bone marrow fibrosis is a hallmark of PMF. Its presence is mandated for the diagnosis of the overt fibrotic stage of PMF.[38] See Criteria.
Aspiration (with a properly placed biopsy needle) usually results in a 'dry tap' in patients with PMF. Biopsy will typically reveal marrow fibrosis (reticulin fibrosis) and megakaryocytic proliferation and atypia. Bone marrow cellularity may be increased, decreased, or hypoplastic. [Figure caption and citation for the preceding image starts]: Bone marrow biopsy showing increased reticulin depositionFrom the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].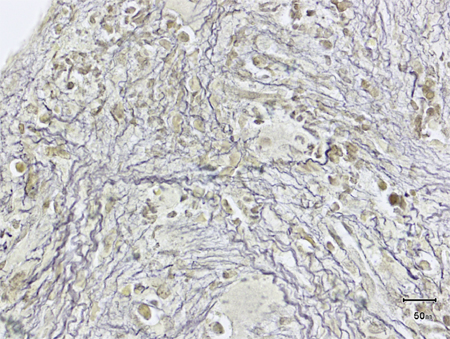 [Figure caption and citation for the preceding image starts]: Trephine bone marrow biopsy showing megakaryocytic hyperplasia and clusteringFrom the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].
[Figure caption and citation for the preceding image starts]: Trephine bone marrow biopsy showing megakaryocytic hyperplasia and clusteringFrom the collection of A. Emadi and J.L. Spivak; used with permission [Citation ends].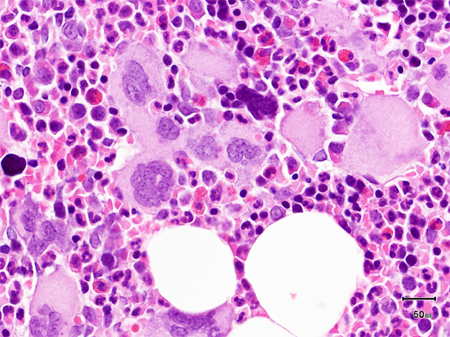
Bone marrow fibrosis alone is not sufficient to diagnose PMF because it can occur in other myeloproliferative neoplasms (MPNs, e.g., PV, ET, and chronic myeloid leukaemia) and other haematological disorders (e.g., systemic mastocytosis, hairy cell leukaemia, myelodysplasia, primary marrow lymphoma, and acute leukaemia). Distinguishing between PV/ET and early 'prefibrotic’ PMF may be difficult based on histological findings, but accurate diagnosis is essential to optimise management.[33]
Confirming the diagnosis, assessing risk and prognosis
Additional tests (e.g., genetic mutation testing, cytogenetic analysis, imaging) may be required to:[26][39]
exclude other disorders that can cause myelofibrosis,
confirm the diagnosis, and
inform risk assessment and prognosis.
Cytogenetic and molecular testing
Fluorescence in situ hybridisation (FISH) or multiplex reverse transcriptase PCR for BCR::ABL1 is required to exclude the diagnosis of chronic myeloid leukaemia.
Somatic driver mutation analysis
All patients with suspected PMF should undergo molecular testing for the JAK2 V617F mutation initially. If negative, testing for MPL and CALR mutations should follow.[26][39] Alternatively, a next-generation sequencing panel comprising all three MPN driver mutations can be used, which also provides quantitative assessment of driver mutation allele burden.
Presence of JAK2 V617F, MPL, or CALR mutation indicates an MPN, but these driver mutations are not specific for PMF. Incidence of JAK2 V617F, MPL, and CALR mutation in PMF patients is reported to be approximately 58%, 8%, and 25%, respectively.[14][15]
Approximately 10% of PMF patients are negative for JAK2, CALR, and MPL mutations (i.e., triple negative); therefore, absence of these MPN driver mutations does not exclude the diagnosis.[24] Some triple-negative patients may have uncommon MPL mutations.[25]
Although MPN driver mutations are not mutually exclusive, patients typically only have one driver mutation that is clonally dominant.
Testing for other (non-driver) genetic mutations (e.g., ASXL1, EZH2, SRSF2, U2AF1 Q157, IDH1/2, SF3B1, TET2) should be carried out for prognostication and risk stratification following diagnosis.[24]
Bone marrow cytogenetic analysis
Chromosomal abnormalities, e.g., involving chromosomes 13 (del.13q), 20 (del.20q), +8 (trisomy 8), 1, 5 (-5/del5q), 7 (-7/del7q), +9 (trisomy 9), 12 (del12p), and 17 are commonly reported in patients with PMF.[12]
Chromosomal abnormalities are not diagnostic for PMF, but their identification can facilitate risk stratification and prognostication. Sole +9 (trisomy 9), 13 (del.13q), 20 (del.20q), and normal cytogenetics have the most favourable prognosis; sole +8 (trisomy 8), 5 (-5/del5q), 7 (-7/del7q), 12 (del12p), 17 (i(17q)), and complex mutations have a distinctly poorer prognosis.[10][11][12][13]
Imaging
Imaging studies (ultrasound, radionuclide, computed tomography [CT], magnetic resonance imaging [MRI], echocardiogram) may be helpful in uncovering extramedullary haematopoiesis. MRI can readily identify spinal extramedullary haematopoiesis, and a technetium 99 (Tc 99m sulfur colloid) scan can uncover pulmonary extramedullary haematopoiesis. Echocardiogram can evaluate the presence of pulmonary hypertension, a manifestation of extramedullary haematopoiesis. Imaging studies should not routinely be used unless extramedullary haematopoiesis is suspected and the site needs to be identified for treatment.
Uric acid level
Hyperuricaemia is a consequence of increased cell turnover and can cause kidney stones or gout, particularly with cytoreductive therapy. Serum uric acid levels may be >416 micromol/L (>7 mg/dL) in men and >357 micromol/L (>6 mg/dL) in women in cases of hyperuricaemia.
Autoimmune phenomena
Autoimmune phenomena are characteristic of PMF.
Testing for autoreactivity can be carried out if clinically indicated (e.g., joint complaints, evidence of haemolysis, or unexplained thrombocytopenia). Results may reveal circulating immune complexes; complement activation; elevated antinuclear antibody, elevated rheumatoid factor titres; and/or a positive Coombs' test in the absence of an overt connective tissue disorder.
Use of this content is subject to our disclaimer