Abordagem
A ataxia se caracteriza pelo movimento desordenado dos membros, tronco e músculos cranianos, em decorrência de uma patologia no cerebelo e respectivas conexões ou nas vias sensoriais proprioceptivas.
A ataxia cerebelar produz um conjunto característico de sinais, revelados pela história e o exame físico:[108][109]
A marcha é instável, com tendência a quedas
A coordenação das mãos é prejudicada
Pode haver presença de disartria ou disfagia
Podem ocorrer sintomas oculares relacionados ao controle anormal do movimento ocular.
Um exame físico neurológico detalhado mostra:
Dismetria dos membros (imprecisão dos movimentos direcionados), como mostra o teste anormal de seguir com o dedo usando os dedos das mãos ou dos pés do paciente
Pode haver presença de tremor cinético e oscilação descontrolada dos membros durante movimentos relativamente lentos, mas direcionados.
Problemas iniciais de postura e marcha, que incluem a incapacidade de permanecer na posição tandem ou com os pés juntos; a base postural torna-se ampla e exibe maior oscilação do corpo. A marcha tandem torna-se prejudicada e, posteriormente, a marcha normal fica claramente atáxica com um caráter cambaleante e de base ampla
O movimento ocular mostra nistagmo induzido pela mirada ou de outros tipos, acompanhamento visual anormal dos objetos apresentados (aparência espasmódica devida ao movimento sacádico de busca) e movimentos sacádicos imprecisos quando a pessoa é solicitada a mover os olhos rapidamente em direção ao alvo (sacadas hipométricas e hipermétricas)
Um tipo escandido de disartria.
Os pacientes com ataxia como principal característica muitas vezes apresentam outros sinais neurológicos não cerebelares, seja porque a lesão causadora da ataxia se estende para além do cerebelo e inclui estruturas adjacentes ou porque a condição degenerativa envolve outras estruturas além do cerebelo.
A ataxia sensorial se caracteriza pela falta de coordenação dos membros superiores e inferiores associada à falta de propriocepção. Os sinais clínicos incluem sensibilidade vibratória prejudicada, além de prejuízo da percepção cinestésica e de posição. Os reflexos tendinosos muitas vezes são ausentes, mas os movimentos oculares e a fala não são afetados. Em alguns pacientes, características sensoriais e cerebelares podem coexistir.
Abordagem em relação ao diagnóstico diferencial
Diante dos diversos distúrbios e da grande variabilidade de apresentação das doenças atáxicas, é útil considerar os pacientes em alguns grupos mais amplos antes de tentar uma abordagem diagnóstica específica.
Os grupos maiores de pacientes são os seguintes:
Pacientes com ataxia aguda
Pacientes com ataxia subaguda ou crônica com anormalidades estruturais definitivas na imagem, com ou sem uma história familiar positiva
Pacientes com ataxia subaguda ou crônica nos quais a imagem mostra atrofia do cerebelo e estruturas associadas (ou, raramente, nenhuma anormalidade), com ou sem uma história familiar.
Os seguintes fatores são modificadores importantes da abordagem diagnóstica:
Idade de início
Evolução da doença
Presença ou ausência de uma história familiar da mesma doença
Se a ataxia é de natureza cerebelar ou sensitiva
Presença de sinais neurológicos não atáxicos
Presença de determinadas características sistêmicas
Presença ou ausência de anormalidade estrutural visível por tomografia computadorizada (TC) ou ressonância nuclear magnética (RNM)
Ataxia aguda
Os pacientes apresentam um início agudo de problemas de equilíbrio, com ou sem sintomas adicionais.
A idade de início é um importante fator determinante da causa. A ataxia de início agudo em crianças provavelmente é resultado de cerebelite aguda ou pode ser induzida por um medicamento (por exemplo, anticonvulsivantes). Em adultos, as causas mais frequentes são AVC hemorrágico ou isquêmico no cerebelo ou tronco encefálico, intoxicações (como medicamentos terapêuticos, álcool e uso indevido de drogas) e síndrome de Wernicke-Korsakoff. Lesões desmielinizantes como esclerose múltipla também podem ter um início rápido e, se ocorrerem no cerebelo ou em suas conexões, apresentarão ataxia aguda. A forma variante de Miller-Fisher da síndrome de Guillain-Barré deve ser considerada.
História
A idade de início é um fator determinante muito importante.
Outras informações fundamentais são a evolução dos sintomas (meses ou alguns anos em contraste com muito crônica), um estudo por imagem do cérebro (preferivelmente ressonância nuclear magnética [RNM]) e uma história familiar de ataxia.
Quando a história familiar é positiva, deve-se determinar, se possível, o padrão de herança (autossômica recessiva, autossômica dominante, ligada ao cromossomo X) a fim de restringir o diagnóstico diferencial.
Obtenha a história de dependência alcoólica e deficit nutricional, bem como a história medicamentosa completa, inclusive qualquer uso indevido de drogas.
Em pacientes com desequilíbrio apenas, uma história de vertigem, zumbido e problemas de audição pode indicar um problema vestibular periférico.
Explore sintomas que indiquem o aumento da pressão intracraniana (por exemplo, cefaleia, náuseas, vômitos) e sintomas que possam indicar problemas nas estruturas contíguas ao cerebelo, como o tronco encefálico. Isso inclui fraqueza ou problemas sensitivos nos membros e deficits dos nervos cranianos. A obnubilação pode ser proeminente com envolvimento do tronco encefálico.
Raramente em crianças e adultos jovens, a apresentação pode anunciar uma doença na qual ocorrem episódios repetidos de ataxia. A história desses episódios e problemas semelhantes em outros membros da família pode indicar uma síndrome de ataxia episódica. Erros inatos do metabolismo também podem apresentar episódios de ataxia.
Exame físico
Ataxia é a falta de coordenação de movimentos intencionais.
Observe oscilações da cabeça e do tronco ou titubeio.
Avalie se a fala apresenta disartria ou é pastosa. Na fala pastosa, o volume da voz do paciente varia de baixo a alto.
Avalie se os movimentos oculares apresentam nistagmo, espasmos na tentativa de rastreio lento e sacadas mais lentas. A paralisia oculomotora pode estar associada a alguns distúrbios.
O nistagmo na posição primária e a ocorrência de "past-pointing" lateralizado podem sugerir uma vestibulopatia aguda, mas é difícil distinguir uma vestibulopatia de um pequeno infarto do tronco encefálico.
Verifique se há rigidez de nuca.
Inclua um exame fundoscópico.
Avalie a marcha verificando a posição de base ampla e a marcha. Teste a marcha em tandem.
Verifique a dismetria dos membros pelos testes dedo-nariz e calcanhar-joelho, a disdiadococinesia e o teste dos movimentos rápidos do calcanhar.
Verifique se há rebote com o teste de toque no punho, no qual o paciente é incapaz de manter a posição depois de um deslocamento inesperado.
A forma variante de Miller-Fisher da síndrome de Guillain-Barré deverá ser considerada se houver ataxia, paralisia oculomotora e arreflexia, com o sem fraqueza adicional nos membros, músculos bulbares e tronco.
Investigações
A investigação com imagens do cérebro (TC, RNM com sequência de imagem ponderada por difusão) é necessária para verificar alterações de sinal associadas a AVC, desmielinização ou cerebelite.[107][110][111][112] A TC em crianças requer o uso de protocolos pediátricos dedicados para minimizar a exposição à radiação.[112][Figure caption and citation for the preceding image starts]: Infarto cerebelar bilateral agudo, observado na ressonância nuclear magnética de sequência de imagem ponderada por difusãoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].
 [Figure caption and citation for the preceding image starts]: Infarto cerebelar observado através de ressonância nuclear magnética (sequência de recuperação da inversão atenuada por fluidos): observe a presença de edema secundário e a obliteração do quarto ventrículoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Infarto cerebelar observado através de ressonância nuclear magnética (sequência de recuperação da inversão atenuada por fluidos): observe a presença de edema secundário e a obliteração do quarto ventrículoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends]. [Figure caption and citation for the preceding image starts]: Infarto medular dorsolateral junto com infartos dispersos no cerebelo, observado na imagem de ressonância nuclear magnética ponderada na sequência de difusãoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Infarto medular dorsolateral junto com infartos dispersos no cerebelo, observado na imagem de ressonância nuclear magnética ponderada na sequência de difusãoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].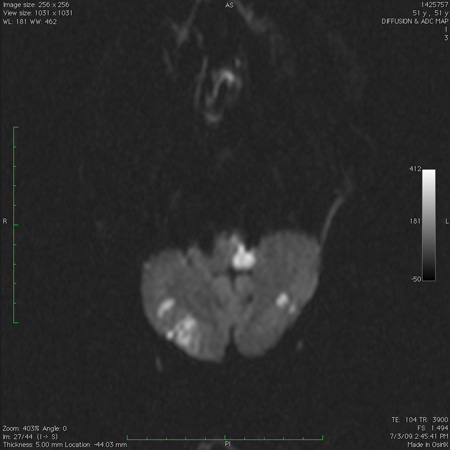 [Figure caption and citation for the preceding image starts]: Tomografia computadorizada do cérebro mostrando hemorragia no cerebelo com extensão para o quarto ventrículoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Tomografia computadorizada do cérebro mostrando hemorragia no cerebelo com extensão para o quarto ventrículoDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].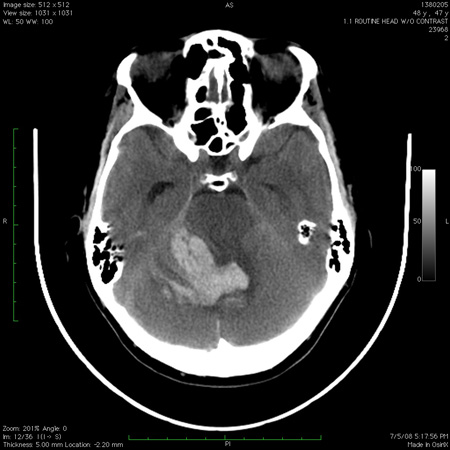
Se o exame de imagem do cérebro constatar um infarto isquêmico no cerebelo ou tronco encefálico, poderá ser necessário realizar agudamente estudos de imagem do sistema vascular em casos específicos; entre eles estão angiotomografia, angiografia por ressonância magnética e angiografia por cateter de rotina.[110]
Deve-se fazer o exame de líquido cefalorraquidiano (LCR) para verificar a presença de parâmetros infecciosos/inflamatórios e a síntese de IgG, caso os achados nos exames de imagem do cérebro:
não consigam confirmar a presença de lesão isquêmica ou hemorragia
sejam compatíveis com esclerose múltipla ou cerebelite.
A síndrome de Wernicke-Korsakoff é diagnosticada principalmente com base clínica e por uma rápida resposta à vitamina B1 e outra suplementação nutricional.
Os pacientes com suspeita de síndrome de Miller-Fisher requerem o exame eletrofisiológico do sistema neuromuscular e o exame de LCR, que pode revelar altos níveis de proteína. O anticorpo para GQ1b muitas vezes é elevado na síndrome de Miller-Fisher, mas não se pode obter os resultados de forma rápida.
Deve-se fazer um rastreamento de uso indevido de substâncias, inclusive abuso de álcool, em pacientes sem lesões estruturais, e medir os níveis adequados de medicamentos naqueles que estejam tomando medicamentos tóxicos para o cerebelo.
Ataxia subaguda ou crônica: anormalidades estruturais definidas nos estudos de imagem
A maioria desses pacientes é adulta e tem uma evolução clínica relativamente mais rápida (medida em meses ou de 1 a 2 anos) que a observada nas formas hereditárias de ataxia ou na ataxia esporádica/idiopática. Além disso, a história familiar de um distúrbio semelhante geralmente é negativa. Os problemas incluem lesões vasculares (residuais de acidente vascular cerebral [AVC] isquêmico/hemorrágico prévio, malformações vasculares), doença desmielinizante (esclerose múltipla), lesões neoplásicas (tumores primários e metastáticos) e lesões compressivas de natureza não neoplásica (isto é, anomalias da junção craniovertebral).
Os pacientes podem ter sinais do sistema nervoso, geralmente de natureza "contígua" (sinais de lesões em estruturas adjacentes como tronco encefálico e parte superior da medula cervical), de aumento de pressão ou sinais em áreas remotas do sistema nervoso como o nervo óptico, os hemisférios e a parte inferior da medula espinhal (este último indica uma doença desmielinizante).
Em qualquer paciente que apresente ataxia crônica/subaguda, deve-se fazer uma ressonância nuclear magnética (RNM) do cérebro com ou sem contraste.[111] A maioria das entidades desse subgrupo pode ser definitivamente diagnosticada com base na história clínica, na natureza dos sinais clínicos e nas anormalidades de imagem. As únicas situações em que esses pacientes podem ter problemas semelhantes na família são alguns tumores familiares raros ou síndromes de malformação como a síndrome de Von Hippel-Lindau.[Figure caption and citation for the preceding image starts]: Lesão de massa grande no cerebelo com efeitos de pressão, conforme observado na ressonância nuclear magnética (RNM)Do acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].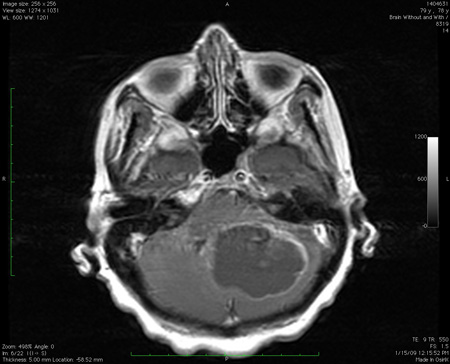
Ataxia subaguda ou crônica: atrofia cerebelar, do tronco encefálico e da medula espinhal em diversas combinações ou nenhuma anormalidade nos estudos de imagem: história familiar negativa
Início na infância
Mesmo quando não há história familiar, essas crianças provavelmente têm uma doença monogênica, já que as diversas causas adquiridas são incomuns em crianças. Por isso, o importante nesse grupo é descobrir uma etiologia genética, seja pela análise do ácido desoxirribonucleico (DNA), pelo achado de um erro inato do metabolismo em teste bioquímico ou por ambos. É necessária uma biópsia de pele e cultura de fibroblastos quando há suspeita de doença de Niemann-Pick tipo C.
A maioria desses distúrbios corresponde a entidades autossômicas recessivas. Outros tipos de doença genética que podem se apresentar nesse grupo são aqueles ligados ao cromossomo X ou de herança mitocondrial. Raramente, uma ataxia autossômica dominante pode se manifestar na infância antes que o genitor afetado se torne sintomático; isso é especialmente provável com a SCA7 e a atrofia dentato-rubro-palido-luisiana (ADRPL). Os detalhes da investigação dessas crianças são apresentados na seção abaixo sobre ataxia infantil com uma história familiar positiva.
Uma forma de ataxia adquirida nas crianças é a síndrome de opsoclonia-mioclonia, que se caracteriza pelo início rápido ou subagudo da ataxia, mioclonias e movimentos oculares caóticos de natureza involuntária. Acredita-se que seja de origem autoimune, mas pode ser desencadeada por uma malignidade subjacente, em geral um neuroblastoma. Estudos de imagem do cérebro e abdome e o exame do líquido cefalorraquidiano (LCR) podem ser úteis.
Início adulto
As causas adquiridas de ataxia predominam nesse grupo e devem ser excluídas prioritariamente.
É um paciente "isolado" adulto típico com ataxia cuja história familiar é negativa para uma doença semelhante. Para se certificar da história familiar negativa, convém fazer uma árvore genealógica com pelo menos 3 gerações e investigar possíveis doenças semelhantes em tantos parentes quanto possível. A idade dos pais na época da morte pode ser importante porque a história familiar pode ser negativa em virtude da morte precoce do genitor afetado.
A anamnese deve explorar abuso de álcool, estado nutricional (por exemplo, perda de peso, doença gastrointestinal crônica), sintomas endócrinos, história de hipóxia significativa ou de intermação, medicamentos administrados atualmente (inclusive medicamentos de venda livre), uso indevido de drogas e presença de doença autoimune específica de um órgão, como diabetes ou hipotireoidismo. Deve-se obter a história ou os fatores de risco de infecção por HIV.
O exame físico deve determinar se a ataxia é de natureza cerebelar ou sensitiva. As ataxias sensitivas são menos comuns e têm um diagnóstico bastante diferencial. A ataxia nesse grupo de pacientes (isto é, com um processo atrófico na imagem) tende a ser simétrica nos dois lados do corpo. A ataxia em pacientes com lesões evidentes detectadas na imagem muitas vezes pode ser assimétrica.
As investigações recomendadas incluem uma RNM do cérebro, que descarte lesões estruturais e documente a presença de atrofia cerebelar, com ou sem atrofia do tronco encefálico, ou da medula espinhal. Estudos da tireoide, níveis de vitamina (isto é, B1, B6, B12), níveis de ácido metilmalônico e homocisteína (que indicam o estado da B12), níveis de medicamentos (por exemplo, fenitoína, carbamazepina, fenobarbital se indicados), anticorpos contra descarboxilase do ácido glutâmico, gliadina e endomísio (transglutaminase tecidual), anticorpos contra células parietais, anticorpos paraneoplásicos, sorologia para HIV, reação em cadeia da polimerase para doença de Whipple, avaliação de picos monoclonais (como imunoeletroforese sérica), anticorpos anti-MAG, biópsia intestinal e eletrofisiologia do nervo periférico, podem contribuir para o diagnóstico. Os testes específicos devem ser selecionados de acordo com as informações clínicas.[Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética (RNM) do cérebro mostrando atrofia cerebelar e do tronco encefálico na SCA 1Do acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].

Se o quadro clínico for duvidoso, deve-se realizar uma pesquisa de câncer visceral como de pulmão, ovário ou mama, incluindo exame físico completo e exames de imagem.
Embora doenças não genéticas dominem esse grupo, em pessoas nas quais não se pode estabelecer nenhum dos diagnósticos acima, deve-se investigar uma mutação mitocondrial, autossômica recessiva ou ligada ao cromossomo X. Em alguns casos, pode-se fazer também uma análise de mutações autossômicas dominantes quando o quadro clínico é preocupante nesse sentido e há dúvida de que a história familiar não é confiável. As anormalidades comuns no DNA que foram observadas em uma série desses pacientes incluem ataxia de Friedreich, síndrome de tremor e ataxia associada ao X frágil, SCA 6, SCA 8 e mutações do DNA mitocondrial.
Além disso, alguns "defeitos congênitos do metabolismo" podem raramente se apresentar nessa faixa etária. Exames laboratoriais que podem indicar alguns desses distúrbios incluem creatina quinase, alfafetoproteína sérica, lactato e níveis de piruvato, níveis de ácidos graxos de cadeia muito longa, níveis de ácido fitânico, quitotriosidase plasmática e níveis de amônia. Entretanto, o rendimento das análises individuais é muito baixo.
Ataxia subaguda ou crônica: atrofia cerebelar, do tronco encefálico e da medula espinhal em diversas combinações ou nenhuma anormalidade nos estudos de imagem: história familiar positiva
Início na infância
A maioria é de doenças autossômicas recessivas, mas as ataxias ligadas ao cromossomo X, mitocondriais e autossômicas dominantes também podem se manifestar na infância. Embora a herança autossômica recessiva seja indicada por irmãos afetados e consanguinidade parental, muitas dessas crianças são pacientes isolados. Muitas ataxias autossômicas recessivas podem ocasionalmente se manifestar na vida adulta e precisam ser consideradas em adultos também.
Raramente, as ataxias autossômicas dominantes podem se manifestar na infância antes que um genitor afetado se torne sintomático (por exemplo, SCA 7 e ADRPL).
Deve-se determinar a idade de início. Algumas entidades como a ataxia com apraxia oculomotora 1 (AOA 1) e a ataxia-telangiectasia (AT) começam na primeira década de vida, enquanto a ataxia de Friedreich e a ataxia com apraxia oculomotora 2 (AOA 2) normalmente, que são mais comuns, se iniciam na segunda década.
Muitos desses casos, incluindo a maioria das crianças com ataxia de Friedreich, têm reflexos tendinosos ausentes, revelando uma doença dos nervos periféricos coexistente. Casos mais raros podem ter espasticidade e reflexos tendinosos vivos associados. Pode-se observar apraxia de movimento ocular (mas não universalmente) na AT, AOA 1 e AOA 2. Também se observa movimentos coreicos associados na AT, AOA 1 e AOA 2, mas não na ataxia de Friedreich. Pode-se notar tremor da cabeça e retinopatia na ataxia por deficiência de vitamina E (ADVE) e retinopatia nas anormalidades do ácido desoxirribonucleico (DNA) mitocondrial. Podem ser observados observar pés cavos na ataxia de Friedreich. Investigue se o paciente apresenta anormalidades sistêmicas.[Figure caption and citation for the preceding image starts]: Deformidade de pés cavos na ataxia de FriedreichDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].

Diferentemente das ataxias autossômicas dominantes, as autossômicas recessivas geralmente revelam acometimento dos tecidos do sistema nervoso não central. Exemplos incluem cardiomiopatia e diabetes na ataxia de Friedreich, telangiectasia cutânea e conjuntival na AT, malignidade linforreticular na AT, alfafetoproteína sérica elevada na AT e AOA 2, hipoalbuminemia na AOA 1, anéis de Kayser-Fleischer e doença hepática na doença de Wilson, e xantomas tendíneos e catarata na xantomatose cerebrotendinosa.[Figure caption and citation for the preceding image starts]: Telangiectasia conjuntival precoce em um paciente com ataxia-telangiectasiaDo acervo de Dr. S. H. Subramony; usado com permissão [Citation ends].
 [Figure caption and citation for the preceding image starts]: Xantoma tendíneo observado na xantomatose cerebrotendinosaDo acervo do Dr. S. H. Subramony, com agradecimento a Dr. Uday Muthane; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Xantoma tendíneo observado na xantomatose cerebrotendinosaDo acervo do Dr. S. H. Subramony, com agradecimento a Dr. Uday Muthane; usado com permissão [Citation ends].
Nessas crianças, deve-se realizar primeiro uma ressonância nuclear magnética (RNM) do cérebro e da parte superior da medula espinhal. Observa-se atrofia na parte alta da medula na ataxia de Friedreich. A atrofia cerebelar é observada na AT, AOA 1 e AOA 2. A eletrofisiologia do nervo periférico pode indicar o envolvimento proeminente dos nervos, observado na ataxia de Friedreich e, em menor grau, na ADVE e nas AOAs.
A mutação autossômica recessiva mais comum é a expansão GAA da AF (restrita a populações indo-europeias) e depois na AOA 2. A AT muitas vezes pode ser clinicamente diagnosticada e confirmada por uma análise de mutação direcionada ou por testes de truncamento de proteínas. Outro teste que se pode empregar é a radiossensibilidade de fibroblastos cultivados.
Como a ADVE pode ser tratada, deve-se obter níveis séricos de tocoferol em crianças e adultos com ataxia crônica de causa indeterminada.
Outras mutações autossômicas recessivas que podem ser obtidas incluem aquelas que causam AOA 1, síndrome de ataxia mitocondrial recessiva, ataxia espástica autossômica recessiva de Charlevoix-Saguenay e ADVE.
Em seguida, deve-se considerar uma anormalidade do DNA mitocondrial. Descarte uma ataxia autossômica dominante através de avaliação cuidadosa da história familiar, exame físico dos pais e análise de mutação (considerações comuns são SCA 7, SCA 2 e ADRPL, sendo que a última é mais comum no Japão), se necessário.
Como os distúrbios genéticos com "defeitos congênitos do metabolismo" geralmente se manifestam nessa faixa etária, essas doenças precisam ser descartadas por estudos metabólicos ou de ácido desoxirribonucleico (DNA). Os exames laboratoriais incluem aminoácidos séricos e na urina, ácidos orgânicos urinários, amônia sérica, testes para doenças do armazenamento lisossomal e colestanol sérico. Outros testes úteis podem incluir espectroscopia por ressonância magnética para detectar leucoencefalopatia com substância branca evanescente, ceruloplasmina sérica, níveis de cobre para a doença de Wilson e níveis de ácido fitânico. Convém realizá-los em colaboração com um especialista em doenças metabólicas.
Início adulto
Em geral, o início nessa faixa etária está associado a uma história familiar consistente com a herança autossômica dominante (SCAs).
Nesse cenário, pode-se realizar um estudo de imagem para descartar uma lesão estrutural coincidente, mas isso talvez não seja necessário se o quadro clínico for totalmente consistente e o médico tiver informações sobre outros membros da família.
Se a mutação na família já for estabelecida, outro membro da família com os sinais clínicos típicos da mesma doença poderá ser diagnosticado exclusivamente por critérios clínicos ou obtendo-se apenas a mutação já conhecida na família. Se a mutação da família for desconhecida, o quadro clínico e o padrão de prevalência de diferentes SCAs poderão ser úteis para determinar o genótipo mais provável, embora todos os especialistas concordem que não se pode fazer um diagnóstico genotípico unicamente com base no fenótipo.
A SCA mais comum em todo o mundo é a SCA3, também conhecida como doença de Machado-Joseph (DMJ). Algumas partes do mundo, como Portugal (especialmente os Açores) e alguns territórios anteriormente ocupados por portugueses, como o Brasil, têm uma prevalência ainda maior de DMJ. Ela é seguida pela SCA2, SCA6 e SCA1, provavelmente nessa ordem, na maioria das regiões do mundo. Todas elas, incluindo a SCA7 (na qual há retinopatia e perda da visão associadas), são causadas por expansões de CAG instáveis em seus respectivos genes e provavelmente devam ser a primeira linha de análise de mutação em um paciente com SCA.
A SCA 1, SCA 2 e SCA 3 podem ter início em vária faixas etárias, mas a idade média de início é a terceira década de vida. A evolução da doença é regular e progressiva, com morte geralmente na quinta década de vida. A SCA 6, no entanto, costuma ter um início tardio na quarta década e uma evolução da doença muitas vezes compatível com a expectativa de vida normal. A SCA 7 e a ADRPL têm início na infância e na vida adulta; a evolução da doença é mais agressiva com o início precoce. A perda da visão na SCA 7 não é universal, mas tende a ocorrer com início precoce.
Uma boa história familiar com detalhes do fenótipo em outros membros da família pode indicar o genótipo. Assim que se exclui as expansões CAG, pode-se considerar uma análise da mutação para a expansão CTG da SCA 8, outros distúrbios de expansão de nucleotídeos como a SCA 12, SCA 10 e SCA 17, e aqueles nos quais se descobriu outros mecanismos mutacionais (por exemplo, SCA 13, SCA 14).
Recentemente, descobriu-se mutações em mais SCAs, mas, em geral, elas só estão disponíveis para teste em laboratórios de pesquisa. É interessante observar que um estudo que utilizou novas técnicas de sequenciamento do exoma em pacientes com apresentações esporádicas e início na fase adulta demonstrou que esse é um teste relevante, oferecendo um diagnóstico definitivo em mais de um quinto dos pacientes e sugerindo um diagnóstico potencial em mais de um terço. Esse estudo postula que, no futuro, o sequenciamento clínico do exoma poderá ser considerado parte da avaliação de rotina de todos os pacientes com ataxia cerebelar progressiva crônica.[113]
A ataxia de início na idade adulta com clara herança autossômica recessiva é menos comum, mas deve ser investigada para crianças com esse tipo de ataxia.
Finalmente, a ataxia de início na idade adulta (geralmente >50 anos) com um processo atrófico em estudos de imagem, sem história familiar, sem mutação estabelecida e com todas as causas adquiridas excluídas, é chamada de ataxia esporádica ou idiopática. Nesse grupo, a detecção de disautonomia por testes detalhados de função autonômica e de denervação do esfíncter anal por eletromiografia pode permitir um diagnóstico de uma provável atrofia de múltiplos sistemas.
Distúrbios semelhantes à ataxia
Muitos tipos de problema de marcha podem se assemelhar superficialmente à ataxia. Os exemplos incluem anormalidades de marcha devidas a lesões dos lobos frontais bilaterais (isto é, ataxia frontal ou de Brun) e, ocasionalmente, marcha distônica. As anormalidades da marcha devido a doença muscular, neuropatias e problemas da medula espinhal em geral podem ser facilmente identificadas pelo exame clínico.
As dificuldades de marcha das lesões do aparelho vestibular também precisam ser diferenciadas da ataxia cerebelar. O problema de coordenação dos membros tem uma natureza mais ligada ao "past-pointing" que à dismetria de fato, e os problemas de movimento ocular geralmente se restringem a nistagmo na posição primária. A disartria não é uma característica de doença vestibular, mas náuseas e oscilopsia podem ser consequências de lesões vestibulares.
A presença de sinais oculomotores e da fala relacionados a uma patologia cerebelar pode claramente diferenciar a ataxia cerebelar de outros tipos de falta de coordenação.
O uso deste conteúdo está sujeito ao nosso aviso legal