Etiology
DiGeorge syndrome (22q11.2 deletion syndrome, or 22q11.2DS) results from a deletion of 1 copy in the 22q11.2 chromosomal region.[17] The deletion results in hemizygosity for at least 30 genes, including TBX1, a key transcription factor for the development of the pharyngeal arches.[2][18] This particular deletion is much more common than other microdeletions due to the genomic structure of this region of chromosome 22.[19] Two areas of low copy number repeats occur at the breakpoints of the deletion; the deletion itself arises when these two areas recombine abnormally during meiosis.[20] The similar structure of the low copy number repeats results in mispairing and a deletion of the portion of the chromosome between the two repeats. The typical deletion is 3 megabases, although 10% to 15% of patients may have a smaller deletion of about 1.5 megabases.[21] This smaller deletion results from abnormal recombination with a third low copy number repeat. Although these deletions are the primary cause of the syndrome, 2% of patients have small atypical 22q11.2 deletions, and a few patients have no deletion.[22][23][24][25][26][27] Phenocopies of DiGeorge syndrome also exist, such as may be found in infants of diabetic mothers.[28] Some of these patients have been found to have point mutations in TBX1, while others have deletions at 10p or mutations in chromodomain-helicase DNA-binding protein.[23][24][25] Genetic variation in TBX1, the principal gene responsible for the syndrome, does not account for the variability in the phenotype seen in the syndrome.[29] There are no known modifiable risk factors for the deletion. Most DiGeorge syndrome cases are sporadic, but known mutations are inherited in an autosomal dominant fashion.[13]
Pathophysiology
DiGeorge syndrome is the set of characteristic morphologic and neurologic features that result from the deletion of 1 copy of 22q11.2.[17] The deletion causes a reduction in TBX1, a key transcription factor for development of the pharyngeal arches.[2]
TBX1 interacts with multiple other signaling molecules, including fibroblast growth factors and vascular endothelial growth factors, to promote pharyngeal arch development.[30] Because TBX1 is insufficient, the pharyngeal arches and pouches are malformed, and the structures they produce are hypoplastic.[31] Haploinsufficiency of TBX1 in mice is sufficient to disrupt the fourth pharyngeal arch artery, replicating a major feature of DiGeorge syndrome.[31] Mice homozygous for TBX1 deletions show complete absence of the thymus and parathyroids, and mice hemizygous for the syntenic deletion show abnormal thymuses.[18][32] TBX1 is also found in the secondary heart field and in the brain, where it may contribute to the development of the neurologic phenotype.[33] Work in mice indicates that when TBX1 is deficient all embryos initially develop features of the disorder, but some seem to be able to correct the defect during later stages of development.[32] This implies that other genes may have a role in modifying the disorder, and might also suggest that development of at least some disease features is preventable.
Work with the mouse model of 22q11.2 deletion has suggested that alterations in microRNA processing caused by deficiency of DGCR8, a gene found in the deleted region, may contribute to the neurologic abnormalities and abnormal brain imaging seen in DiGeorge syndrome.[34][35]
Polymorphisms in some genes have been identified that seem to increase the risk of specific complications.[36][37][38]
Polymorphisms in the catechol-O-methyltransferase gene in the deleted region may increase the risk of schizophrenia, although studies are conflicting. Vascular endothelial growth factor polymorphisms alter the incidence of heart disease in a murine model of the disorder. Similar mechanisms may be at work in humans. These polymorphisms are not yet available for clinical testing or predicting outcomes for patients.
Autoimmunity is also a feature of 22q11.2 deletion and is correlated with the severity of T-cell lymphopenia.[39][Figure caption and citation for the preceding image starts]: Loss of 1 copy of 22q11.2, demonstrated by microarray copy number analysisFrom the collections of Sean A. McGhee, MD and Maria Garcia Lloret, MD [Citation ends].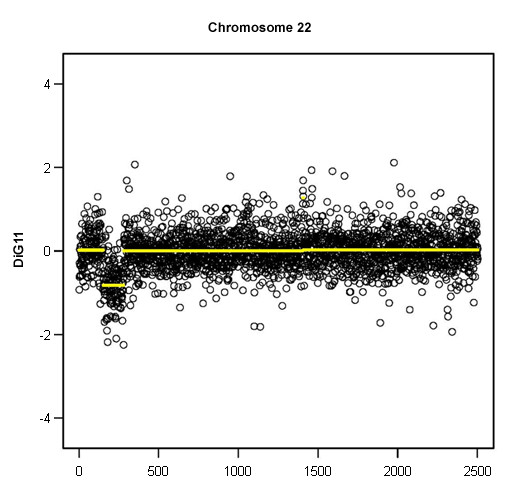
Classification
Complete and partial DiGeorge syndrome (ESID/PAGID diagnostic criteria)[4]
DiGeorge syndrome has been historically divided into complete and partial DiGeorge syndrome, although a greater appreciation of the syndrome's variability has made this distinction less useful.
Complete DiGeorge syndrome was used if patients had the full spectrum of typical manifestations, including severe immunodeficiency.
Partial DiGeorge syndrome was used if patients had only some manifestations of the disorder, particularly those without evident immunodeficiency. Partial DiGeorge syndrome is far more common than complete.
Measurement of naive T-cell numbers is key to distinguishing partial from complete DiGeorge syndrome.[5]
Typical and atypical DiGeorge syndrome[6]
Observations have identified a small subset of patients with DiGeorge syndrome who have T-cell immunodeficiency but develop abnormal oligoclonal T cells, resulting in a graft-versus-host disease-like rash and requiring immunosuppression to avoid complications of lymphoproliferation.[6]
Typical DiGeorge syndrome is used when patients have varying degrees of immunodeficiency but no oligoclonal T-cell populations.
Atypical DiGeorge syndrome is used when patients have proliferating autoreactive oligoclonal T-cell populations.
Both of these distinctions (typical and atypical) apply only to patients with complete DiGeorge syndrome, which specifically refers to severe T-cell immunodeficiency (athymia).
DiGeorge syndrome can also be associated with significant B-cell abnormalities in a higher proportion of patients than previously thought.[7][8]
Use of this content is subject to our disclaimer