Etiology
Genetic mutations identified in 17 genes account for congenital LQTS, with those in the following 3 genes constituting 90% to 95% of cases where a gene can be identified.[3][5][8][16][19][20]
LQT1 arises from loss-of-function mutations in the KCNQ1 gene, which encodes a potassium channel responsible for the slow component of the delayed rectifier current (IKs). A homozygous mutation in KCNQ1 results in the autosomal recessive Jervell and Lange-Nielsen syndrome (JLNS).
LQT2 arises from loss-of-function mutations in the KCNH2 gene, which encodes a potassium channel responsible for the rapid component of the delayed rectifier current (IKr).
LQT3 arises from gain-of-function mutations in the SCN5A gene, which encodes a sodium channel.
While the number of genes implicated in causing LQTS has expanded to 17, some have limited or disputed evidence casting doubt on their association with disease.[7]
Acquired LQTS results from a wide variety of causative factors:
Some of the drugs known to prolong the QT interval or cause depletion of potassium and/or magnesium are quinidine, procainamide, sotalol, amiodarone, disopyramide, dofetilide, phenothiazines, tricyclic antidepressants, and methadone.[21][22] Credible Meds (Arizona CERT): drugs that prolong the QT interval Opens in new window Certain cancer treatments are also known to cause QT prolongation, e.g., kinase inhibitors, growth factor inhibitors, androgen-deprivation therapies, and chimeric antigen receptor T-cell therapies.[23][24]
Electrolyte imbalances: in particular hypokalemia, hypomagnesemia, and hypocalcemia.
Bradyarrhythmias: any sudden bradycardia or atrioventricular (AV) nodal block may result in QT prolongation or pause-dependent QT prolongation.
Central nervous system (CNS) lesions: such as intracranial hemorrhage (especially subarachnoid hemorrhage) and ischemic strokes.
Malnutrition: liquid protein diet, starvation.
Intense exercise training may be a cause of reversible long QT interval in athletes.[25]
Pathophysiology
In congenital LQTS, a number of identified genetic mutations cause the alteration of a specific ion channel current, leading to the pathophysiologic prolongation of repolarization, which equates to QT interval prolongation on the ECG.
In LQT1 and LQT2, mutations reduce function of the delayed rectifier potassium currents (IKs and IKr, respectively), which shuttle potassium ions out of the myocardial cell during repolarization, thus making the cell more negative and returning it to the baseline state of approximately -90 mV.
LQT1 results from heterozygous loss-of-function mutations in the KCNQ1 gene, which encodes the alpha subunit of the slow-activating potassium channel responsible for the slow component of IKs. These mutations lead to dysfunctional IKs channels, which in turn lead to dispersion of repolarization from the epicardial to the endocardial surface, allowing potential development of ventricular tachyarrhythmias. Electrocardiographically, there are characteristic prolonged QT intervals associated with a broad-based T wave.[16][Figure caption and citation for the preceding image starts]: ECG findings in type 1 long QT syndromeFrom the collection of Dr James P. Daubert [Citation ends].
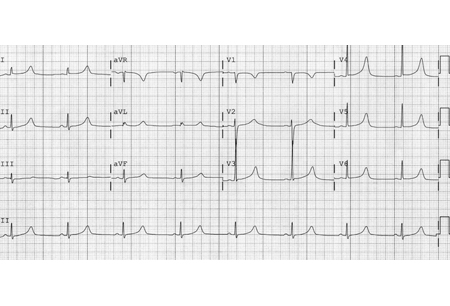
LQT2 results from mutations in the KCNH2 gene, which encodes the alpha (HERG) subunit of the potassium channel responsible for the rapid component of IKr. These mutations lead to dysfunctional IKr channels, which in turn lead to slowed repolarization and transmural dispersion of repolarization. This predisposes to ventricular tachyarrhythmias, particularly torsades de pointes. Electrocardiographically, there are characteristic low-amplitude and notched T waves.[16][Figure caption and citation for the preceding image starts]: ECG findings in type 2 long QT syndromeFrom the collection of Dr James P. Daubert [Citation ends].

LQT3 results from mutations in the SCN5A gene, which encodes for rapidly inactivating sodium channels, resulting in a gain of function of a late sodium current allowing an inward flow of sodium ions to persist long into the plateau phase of the action potential, thereby prolonging repolarization. Electrocardiographically, there are characteristic long ST segments with a late-appearing T wave resulting in a long QT interval.[16][Figure caption and citation for the preceding image starts]: ECG findings in type 3 long QT syndromeFrom the collection of Dr James P. Daubert [Citation ends].
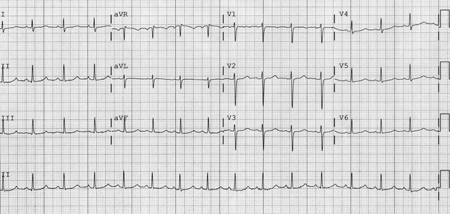
A hereditary form of complete AV block resulting from degeneration of the bundle of His and its branches has been linked to the SCN5A gene (mutations of which are responsible for LQT3), which encodes for rapidly inactivating sodium channels, resulting in a gain of function of a late sodium current allowing an inward flow of sodium ions to persist long into the plateau phase of the action potential, thereby prolonging repolarization.
The many causes of acquired LQTS result in prolongation of the QT interval through a variety of pathophysiologic mechanisms.[26]
Hypokalemia causes hyperpolarization of myocardial cell membranes with consequent prolongation of repolarization, which equates to QT interval prolongation on the ECG.
Hypomagnesemia, which often coexists with hypokalemia, causes early afterdepolarizations, which in turn lead to prolonged repolarization of myocardial cells and subsequent QT interval prolongation on the ECG.
Hypocalcemia prolongs the plateau phase of the action potential, thereby prolonging repolarization of myocardial cells, which equates to prolongation of the QT interval on the ECG.
The likelihood of developing acquired LQTS is influenced by genetic variants that affect the repolarization reserve of a patient, such as D85N in KCNE1.[5]
Classification
Congenital
The condition is inherited as a monogenic disorder with primarily autosomal dominant inheritance and variable penetrance. Multiple genetic mutations have been identified as the cause of LQTS.[3][4][5][6]
LQT1 is due to mutations in the KCNQ1 gene.
LQT2 is due to mutations in the KCNH2 gene.
LQT3 is due to mutations in the SCN5A gene.
LQT4 to LQT17 have been described but are responsible for <10% of cases; affected genes include CALM1/2/3, TRDN, KCNE1/2, KCNJ2, and CACNA1C.
While 17 genes have been implicated in causing LQTS, some have limited or disputed evidence casting doubt on their association with disease.[7]
Inherited as either an autosomal dominant or a recessive trait, LQTS can be phenotypically classified into several congenital syndromes.
Romano-Ward syndrome is inherited as an autosomal dominant trait. It may result from a mutation in any one of the identified genes and is not associated with deafness.[8]
Jervell and Lange-Nielsen syndrome is inherited as an autosomal recessive trait and results from a homozygous mutation in KCNQ1. It is clinically characterized by a very severe form of LQTS and sensorineural deafness,[9] and those affected may experience their first cardiac event during infancy. This is much rarer than the Romano-Ward (autosomal dominant) pattern.
Andersen-Tawil syndrome, also known as hypokalemic periodic paralysis or LQT7, is a rare autosomal dominant condition resulting from a mutation in the KCNJ2 gene.[5] These patients have periodic paralysis and ventricular tachyarrhythmias, and have a variety of dysmorphic features.[10]
Timothy syndrome (LQTS8), a rare autosomal dominant syndrome associated with syndactyly, cardiac malformations, autism, and dysmorphic features.[2]
Acquired
Many factors are associated with the development of a prolonged QT interval:
Drugs
Electrolyte imbalances
Bradyarrhythmias
CNS lesions
Malnutrition
Pathologic genetic variants in KCNE1 and KCNE2.[5]
Use of this content is subject to our disclaimer