Approach
Physical findings
The diagnosis of epidermolysis bullosa (EB) is suggested at birth or during early infancy by the presence of mechanical fragility of the skin, recurrent blisters and erosions, and poorly healing wounds. Additional cutaneous clues include the presence of milia, atrophic scarring, dystrophic or absent nails, herpetiform blistering (seen exclusively in severe EB simplex [EBS]), reticulate hyperpigmentation (EBS with mottled pigmentation [EBS-MP]), and/or exuberant granulation tissue (severe junctional EB [JEB]). Presentation of these skin signs varies according to the EB subtype. Onset in mid or late childhood is more indicative of localized EBS, or late-onset JEB.
Resolution of blistering within the first 1 to 2 years of life is characteristic of bullous dermolysis of the newborn, a rare subtype of both types of dystrophic EB.[34]
Common distributions of lesions on the skin include both generalized and localized patterns (e.g., palms and soles in localized EBS). Inverse (i.e., intertriginous), acral, and centripetal distributions occur less commonly. By definition, recessive dystrophic EB (RDEB), inversa and JEB, inversa have inverse distributions, but until the first year of life may instead appear with generalized skin involvement. Similarly, centripetal distribution is seen only in RDEB, centripetalis, but it too can appear more generalized in distribution until after the first 1 to 2 years of life.[35]
EB subtypes may be further suggested by the presence of specific extracutaneous findings including muscular dystrophy in EBS-MD attributable to underlying PLEC mutations, enamel hypoplasia of the teeth (all JEB subtypes), pyloric atresia (EBS-PA and JEB-PA), severe upper airway disease (primarily in generalized forms of JEB and laryngo-onycho-cutaneous syndrome [JEB-LOC]), tracheolaryngeal stenosis or stricture (in JEB, both severe and intermediate), and pseudosyndactyly (in RDEB more commonly than dominant dystrophic EB [DDEB] and JEB). Some of these complications may not occur until late childhood or adulthood, preventing accurate subclassification on the basis of clinical findings in some neonates and infants.[9][24][25][Figure caption and citation for the preceding image starts]: Axillary contractions and granulation tissue in a child with junctional EB, generalized severe (JEB-gen sev)From the collection of J.D. Fine, MD [Citation ends].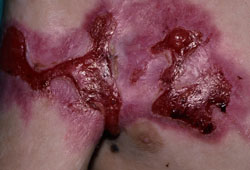
Identification of risk factors
The only known risk factor for EB is a positive family history. Most patients with confirmed autosomal dominant forms of EB have a family history of the disease occurring in a parent. Incomplete penetrance is very uncommon in autosomal dominant EB.
Investigations
Given the considerable overlap in clinical findings among many of the EB types and subtypes, immunofluorescence antigen mapping (IFM) and/or transmission electron microscopy (EM) was typically undertaken in every patient newly diagnosed with EB, unless diagnosis and classification had been previously confirmed on another affected family member. Because far fewer laboratories are experienced in performing and interpreting transmission EM on EB skin than IFM, and because EM is far more expensive and labor intensive to perform, IFM is the preferred method, if both tests cannot be obtained.[36]
However, with the increasing availability, speed, and concurrent diminishing cost of DNA mutational analyses, the investigational paradigm has shifted and clinicians in specialized centers are now able to access EB-specific and skin fragility gene panels utilizing targetted or whole-exome sequencing, facilitating rapid and efficient diagnosis in suspected EB.[8][36] These may now be undertaken as first-line investigations.
IFM, performed on cryopreserved skin specimens from freshly induced blisters, determines the exact level of skin cleavage, by staining with antibodies to specific basement membrane proteins. When additional monoclonal antibodies are employed, IFM helps to distinguish severe JEB from intermediate JEB and DDEB from RDEB, as well as to identify cases of EBS-MD, bullous dermolysis of the newborn, and Kindler EB. It is as accurate as EM at distinguishing among EBS, JEB, DDEB, and RDEB.[3][9][37]
Transmission EM is performed on skin harvested from a fresh, spontaneous blister, or from one induced by traction. This allows direct visualization of the ultrastructural level within which blisters form, separating patients into those having intraepidermal (EBS), intralamina lucida (JEB), sublamina densa (DDEB, RDEB), and mixed (Kindler EB) levels of skin cleavage. Semiquantification of specific structures (hemidesmosomes; anchoring fibrils) can further suggest specific EB subtypes, although there is considerable overlap between some subtypes, reducing the sensitivity and specificity of these laboratory findings when utilized in isolation.[1][3][9][37]
DNA mutational analysis will confirm the type of EB present, as well as the genetic mode of transmission.[3][9] Genotype-phenotype correlation remains poorly resolved within certain subtypes; however, DNA sequencing may not be available to all patients nor accessible to all clinicians. DNA testing is routinely recommended when prenatal diagnosis is being contemplated, or for use in preimplantation in vitro fertilization. If there are existing genetic test results, do not perform repeat testing unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[38]
Routine light microscopy is not recommended as part of the conventional diagnostic workup given the low sensitivity and specificity in distinguishing between even the 4 major EB types by routine staining techniques.
Specialist referral for diagnosis
Any patient with inherited EB, particularly newborns, must be evaluated by a dermatologist experienced in the diagnosis and management of this disease. Where there is any doubt over exact diagnosis, referral to a specialized EB center (or academic dermatology center if the former is unavailable) is recommended. People with extracutaneous presenting features are referred to medical or surgical specialists skilled in the evaluation and subsequent treatment of those particular outcomes.
Use of this content is subject to our disclaimer