Approach
History
Almost all affected people are asymptomatic for much of their lives.[2][28]
A few patients with class I variants have longstanding symptoms: they may have a family history (with a characteristic X-linked pedigree), typically describe neonatal jaundice, and usually give a history of lack of energy and chronic fatigue, pallor, and jaundice and often of having received a blood transfusion in childhood. They may have exacerbations of their symptoms at times of infection, may have increased susceptibility to infection (sometimes even bleeding, due to platelet dysfunction), and may have a premature cataract. They may have abdominal pain and other symptoms of gallstones and gallbladder disease as a consequence of chronic hemolytic anemia.[29]
A detailed history should be elicited, including diet and medications, particularly if the presentation is acute. There is typically a history of the precipitating event over the preceding 1 to 2 days: for example, of ingesting an implicated oxidative drug or foodstuff (e.g., broad beans).[Figure caption and citation for the preceding image starts]: Drugs and agents that are known to cause hemolysis in people with G6PD deficiencyCreated by Professor Atul B. Mehta [Citation ends].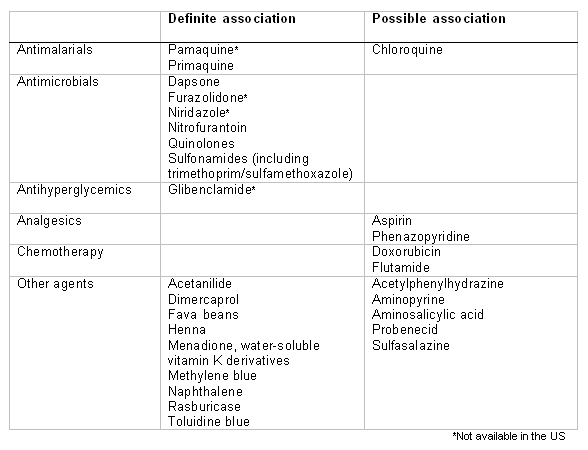 Significant intercurrent infection is also a precipitant.
Significant intercurrent infection is also a precipitant.
Favism (following exposure to broad beans) and drug-induced hemolysis are usually both intravascular and extravascular. Intravascular hemolysis is usually associated with pronounced clinical symptoms such as severe vomiting, headache, prostration, back pain, and the passage of dark urine. Extravascular hemolysis causes less dramatic symptoms (for example, tiredness, jaundice, and listlessness) and can be associated with splenomegaly, although in practice this can take many days to develop and is seen only in people with class I variants.
Physical examination
Pallor and jaundice are the cardinal signs.
Tachycardia and signs of a hyperdynamic circulation are often present with moderate to severe anemia. There may even be an altered level of consciousness. Splenomegaly is usually not detected, but may be present in patients with chronic extravascular hemolysis.
Laboratory tests
The initial tests to perform are CBC and reticulocyte count, with review of the peripheral blood smear, complete metabolic panel (CMP), and urinalysis.
CBC and reticulocytes: show anemia (Hb 4-10 g/dL, depending on severity) with an increased MCHC. The reticulocyte count is typically markedly elevated (>10%).
Peripheral smear: shows characteristic changes of anisocytosis with "bite" cells. Heinz bodies (fragments of denatured Hb) are seen in acute hemolysis. Spherocytes are not a prominent feature in this condition.
CMP: liver function tests show elevated unconjugated bilirubin and markedly elevated lactate dehydrogenase as a consequence of RBC destruction and hemoglobin release. Transaminase levels are elevated and renal function may be abnormal due to dehydration. Hemoglobinuria due to intravascular hemolysis can also cause acute renal failure. Haptoglobin may be requested and is low because of intravascular hemolysis.
Urinalysis: shows hemoglobinuria if intravascular hemolysis is present. Dipstick testing detects urobilinogen and protein.
Imaging
Abdominal ultrasound may be useful for evaluating splenomegaly. Gallstones may be present in those with chronic hemolysis.
Specific tests for G6PD deficiency
The screening test for G6PD deficiency uses a fluorescent spot test.[25] In the test, glucose-6-phosphate is oxidized by G6PD to generate 6-phosphogluconolactone, with concomitant reduction of NADP (nicotinamide adenine dinucleotide phosphate) to NADPH (reduced nicotinamide adenine dinucleotide phosphate). NADPH fluoresces when excited with 340 nm light; failure of the blood spot to fluoresce under UV light is indicative of G6PD deficiency.
The dye decolorization test is an alternative test; the rate of decolorization of the dye by NADPH gives a measure of the activity of G6PD.[25]
Reticulocytes have a higher activity than mature red cells and, in the immediate aftermath of a hemolytic episode, reticulocytosis can elevate the measured activity level to within the normal range. All patients should be retested following a hemolytic episode of unknown cause to ensure that G6PD deficiency is not missed. Final G6PD activity should be interpreted in light of the reticulocyte count measured on the same sample.[25]
Quantitative assay
Quantitative spectrophotometric assay should be undertaken to confirm or exclude diagnosis if the screening test is abnormal or borderline.[25][30][31]
Screening tests can not reliably diagnose G6PD deficiency in heterozygous females.[25] Therefore, G6PD activity should be measured by quantitative spectrophotometric assay first line in female patients. A cytochemical test should be undertaken if quantitative assay results are intermediate or equivocal or if there is a clinical or genetic reason to suspect that a woman is heterozygous for G6PD deficiency.[25][30][31]
Point of care tests
Rapid diagnostic testing can be useful in certain situations at the point of care (e.g., neonatal screening programs or to determine the safety of primaquine treatment in the context ofPlasmodium vivax malaria control programs), but availability may be limited.[25][32] This should be followed by definitive quantitative assay of G6PD activity by spectrophotometry.
Molecular diagnosis
Molecular analysis can be considered when initial tests are equivocal. Candidates for molecular analysis include heterozygous females, and males with Klinefelter syndrome (who may have intermediate G6PD activity).[25]
G6PD variants can be ascertained using polymerase chain reaction (PCR).[33][34][35] If there are existing genetic test results, do not perform repeat testing unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[36]
Use of this content is subject to our disclaimer