Diagnosis is typically made in childhood after presentation with recurrent bacterial and/or fungal infections.
Key history
As CGD patients' mechanism to kill bacteria and fungi is defective, patients often present with a history of recurrent bacterial or fungal infections.
These may include:
pneumonia
soft tissue abscesses
internal abscesses (especially hepatic)
oral infections
osteomyelitis
adenitis
BCGosis
gastrointestinal (GI) infections or obstruction (secondary to granuloma formation)
genitourinary infections or obstruction (secondary to granuloma formation) with symptoms including urgency, flank pain, poor urine stream, pain on urination, bloody urine
chronic nasal infections
skin infections (i.e., furuncles, impetigo, eczema complicated by infection, perianal abscesses)
fatigue
symptoms of colitis or other intestinal inflammation (i.e., diarrhea that is sometimes bloody, abdominal pain, nausea, and vomiting).
A history of infection with Staphylococcus aureus, Aspergillus species, Nocardia species, Serratia marcescens, or Burkholderia cepacia is most common and suspicious.[1]Rosenzweig SD, Uzel G, Holland SM. Phagocyte disorders. In: Stiehm ER, Ochs HD, Winkelstein JA, eds. Immunologic disorders in infants and children. Philadelphia, PA: Elsevier Saunders; 2004:618-51.[3]Winkelstein JA, Marino MC, Johnston RB Jr., et al. Chronic granulomatous disease: report on a national registry of 368 patients. Medicine (Baltimore). 2000 May;79(3):155-69.
http://www.ncbi.nlm.nih.gov/pubmed/10844935?tool=bestpractice.com
[4]van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668749/?tool=pubmed
http://www.ncbi.nlm.nih.gov/pubmed/19381301?tool=bestpractice.com
[31]Marciano BE, Spalding C, Fitzgerald A, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015 Apr 15;60(8):1176-83.
http://www.ncbi.nlm.nih.gov/pubmed/25537876?tool=bestpractice.com
Patients do not generally show particularly increased susceptibility to Candida species, although these organisms have occasionally been implicated as a cause of death.[32]Henriet S, Verweij PE, Holland SM, et al. Invasive fungal infections in patients with chronic granulomatous disease. Adv Exp Med Biol. 2012 Dec 11;764:27-55.
http://www.ncbi.nlm.nih.gov/pubmed/23654055?tool=bestpractice.com
[31]Marciano BE, Spalding C, Fitzgerald A, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015 Apr 15;60(8):1176-83.
http://www.ncbi.nlm.nih.gov/pubmed/25537876?tool=bestpractice.com
Disseminated infection after BCG vaccination is well described, and tuberculosis is not uncommon, especially in endemic areas.[33]Conti F, Lugo-Reyes SO, Blancas Galicia L, et al. Mycobacterial disease in patients with chronic granulomatous disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016 Jul;138(1):241-8.
http://www.ncbi.nlm.nih.gov/pubmed/26936803?tool=bestpractice.com
Other history may include chronic colitis, sometimes misdiagnosed as Crohn disease, or a history of GI or urinary tract obstruction due to granuloma formation.[4]van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668749/?tool=pubmed
http://www.ncbi.nlm.nih.gov/pubmed/19381301?tool=bestpractice.com
[6]Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004 Aug;114(2):462-8.
http://www.ncbi.nlm.nih.gov/pubmed/15286231?tool=bestpractice.com
[7]Huang JS, Noack D, Rae J, et al. Chronic granulomatous disease caused by a deficiency of p47phox mimicking Crohn's disease. Clin Gastroenterol Hepatol. 2004 Aug;2(8):690-5.
http://www.ncbi.nlm.nih.gov/pubmed/15290662?tool=bestpractice.com
[8]Huang A, Abbasakoor F, Vaizey CJ. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis. 2004 Aug;2(8):690-5.
http://www.ncbi.nlm.nih.gov/pubmed/16970572?tool=bestpractice.com
[9]Schappi MG, Smith VV, Goldblatt D, et al. Colitis in chronic granulomatous disease. Arch Dis Child. 2001 Feb;84(2):147-51.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1718666/pdf/v084p00147.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11159292?tool=bestpractice.com
[10]Schappi MG, Klein NJ, Lindley KJ, et al. The nature of colitis in chronic granulomatous disease. J Pediatr Gastroenterol Nutr. 2003 May;36(5):623-31.
https://www.jpgn.org/pt/re/jpgn/fulltext.00005176-200305000-00006.htm
http://www.ncbi.nlm.nih.gov/pubmed/12717086?tool=bestpractice.com
[11]Walther MM, Malech H, Berman A, et al. The urological manifestations of chronic granulomatous disease. J Urol. 1992 May;147(5):1314-8.
http://www.ncbi.nlm.nih.gov/pubmed/1569675?tool=bestpractice.com
Of note, skin infections are often frequent and difficult to treat. Chronic lung disease is increasingly recognized in adulthood, including inflammatory lesions, bronchiectasis, scarring, and emphysema.[34]Campos LC, Di Colo G, Dattani V, et al. Long-term outcomes for adults with chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol. 2021 Mar;147(3):1104-7.
https://www.doi.org/10.1016/j.jaci.2020.08.034
http://www.ncbi.nlm.nih.gov/pubmed/32971110?tool=bestpractice.com
[35]Salvator H, Mahlaoui N, Catherinot E, et al. Pulmonary manifestations in adult patients with chronic granulomatous disease. Eur Respir J. 2015 Jun;45(6):1613-23.
http://www.ncbi.nlm.nih.gov/pubmed/25614174?tool=bestpractice.com
Patients are typically male and show signs and symptoms of disease prior to age 5. There is often a positive family history of either diagnosis with CGD or recurrent, multisystemic infections.
Patients often show mild symptoms despite the presence of serious infection.
Female patients who are carriers of gp91phox mutations often have oral ulcers, a history of discoid lupus, skin rashes, and joint pain.[36]Rosenzweig SD. Inflammatory manifestations in chronic granulomatous disease (CGD). J Clin Immunol. 2008 May;28 Suppl 1:S67-72.
http://www.ncbi.nlm.nih.gov/pubmed/18193341?tool=bestpractice.com
They also frequently describe gastrointestinal symptoms and some may have inflammatory bowel disease.[37]Battersby AC, Braggins H, Pearce MS, et al. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol. 2017 Aug;140(2):628-30.
http://www.ncbi.nlm.nih.gov/pubmed/28343844?tool=bestpractice.com
Rarely, female patients may present with typical sequelae (infection and inflammation) of X-CGD as a result of skewed X-inactivation patterns.[38]Marciano BE, Zerbe CS, Falcone EL, et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J Allergy Clin Immunol. 2018 Jan;141(1):365-71.
http://www.ncbi.nlm.nih.gov/pubmed/28528201?tool=bestpractice.com
Patients with p47phox deficiency may have a milder clinical course than other subtypes, but this is not necessarily the case.
Key physical exam findings
Patients often present with infection. Examination may reveal signs of infection, although patients may be asymptomatic or only mildly ill-appearing despite serious infection.[39]Liese J, Kloos S, Jendrossek V, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr. 2000 Nov;137(5):687-93.
http://www.ncbi.nlm.nih.gov/pubmed/11060536?tool=bestpractice.com
[40]Finn A, Hadzic N, Morgan G, et al. Prognosis of chronic granulomatous disease. Arch Dis Child. 1990 Sep;65(9):942-5.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1792120/?pageindex=1
http://www.ncbi.nlm.nih.gov/pubmed/2221966?tool=bestpractice.com
[41]Fischer A, Segal AW, Seger R, et al. The management of chronic granulomatous disease. Eur J Pediatr. 1993 Nov;152(11):896-9.
http://www.ncbi.nlm.nih.gov/pubmed/8276018?tool=bestpractice.com
In the absence of infection, general physical exam findings may suggest chronic illness, such as poor growth and chronic lymphadenopathy and hepatosplenomegaly.[39]Liese J, Kloos S, Jendrossek V, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr. 2000 Nov;137(5):687-93.
http://www.ncbi.nlm.nih.gov/pubmed/11060536?tool=bestpractice.com
[40]Finn A, Hadzic N, Morgan G, et al. Prognosis of chronic granulomatous disease. Arch Dis Child. 1990 Sep;65(9):942-5.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1792120/?pageindex=1
http://www.ncbi.nlm.nih.gov/pubmed/2221966?tool=bestpractice.com
Signs may include:
fever
clinical findings of pneumonia (i.e., cough, fever, dyspnea, abnormal lung examination)
overt signs of cutaneous infection, adenitis, perianal abscess, or soft tissue abscess (i.e., red skin lesion, perianal pain)
signs of oral infection
skin scarring related to previous abscess drainage or biopsies
chorioretinal lesions
hepatomegaly, splenomegaly.
Routine labs and imaging
Tests include routine laboratory tests (CBC, ESR, and CRP) and imaging (CT, ultrasound, PET scan), along with procedures to identify the infectious agent. The diagnostics currently used for invasive aspergillosis have low sensitivity in CGD patients.[32]Henriet S, Verweij PE, Holland SM, et al. Invasive fungal infections in patients with chronic granulomatous disease. Adv Exp Med Biol. 2012 Dec 11;764:27-55.
http://www.ncbi.nlm.nih.gov/pubmed/23654055?tool=bestpractice.com
Chronic anemia is common (low Hb, low Hct), and ESR and CRP may be elevated during infection. Inflammatory markers may be misleading as they tend to rise and fall in relation to chronic inflammatory processes, even in the absence of infection.
During active infection, prompt imaging, such as CT or ultrasound imaging, is useful to localize and evaluate the extent of infection. Whole body F-18 fluorodeoxyglucose (FDG) PET is helpful in differentiating active infectious processes from past infections or chronic inflammatory lesions.[42]Gungor T, Engel-Bicik I, Eich G, et al. Diagnostic and therapeutic impact of whole body positron emission tomography using fluorine-18-fluoro-2-deoxy-D-glucose in children with chronic granulomatous disease. Arch Dis Child. 2001 Oct;85(4):341-5.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1718940/pdf/v085p00341.pdf
http://www.ncbi.nlm.nih.gov/pubmed/11567949?tool=bestpractice.com
Endoscopy, especially colonoscopy, may be required to evaluate the activity of inflammatory colitis. MRI is useful in cases of suspected osteomyelitis. Brain imaging with MRI should be considered in patients with disseminated Aspergillus or Nocardia infections.[43]Thomsen IP, Smith MA, Holland SM, et al. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J Allergy Clin Immunol Pract. 2016 Nov-Dec;4(6):1082-8.
http://www.ncbi.nlm.nih.gov/pubmed/27178966?tool=bestpractice.com
With increasing recognition of chronic lung disease in CGD, many clinicians now perform routine chest CT scans, for example every 5 years. Abnormal lung function tests can point toward chronic lung disease.[35]Salvator H, Mahlaoui N, Catherinot E, et al. Pulmonary manifestations in adult patients with chronic granulomatous disease. Eur Respir J. 2015 Jun;45(6):1613-23.
http://www.ncbi.nlm.nih.gov/pubmed/25614174?tool=bestpractice.com
Fecal calprotectin levels correlate with colitis severity and can be used to monitor activity.[44]Lowe DM, Smith PJ, Moreira F, et al. Chronic granulomatous disorder-associated colitis can be accurately evaluated with MRI scans and fecal calprotectin level. J Clin Immunol. 2019 Jul;39(5):494-504.
https://www.doi.org/10.1007/s10875-019-00651-2
http://www.ncbi.nlm.nih.gov/pubmed/31172380?tool=bestpractice.com
Confirmatory testing
When suspected in patients with a consistent medical history, referral to a specialist is imperative for diagnostic testing. Diagnosis is typically made by identifying abnormal neutrophil oxidative burst activity. The first of two common methods is the nitroblue tetrazolium (NBT) test, in which normal neutrophils reduce nitroblue tetrazolium to formazan, a dark blue pigment visible on microscopic inspection.[45]Repine JE, Rasmussen B, White JG. An improved nitroblue tetrazolium test using phorbol myristate acetate-coated coverslips. Am J Clin Pathol. 1979 May;71(5):582-5.
http://www.ncbi.nlm.nih.gov/pubmed/453076?tool=bestpractice.com
[46]Johansen KS. Nitroblue tetrazolium slide test. Use of the phorbol-myristate-acetate-stimulated NBT-reduction slide test for routine and prenatal detection of chronic granulomatous disease and diagnosis of heterozygous carriers. Acta Pathol Microbiol Immunol Scand [C]. 1983 Dec;91(6):349-54.
http://www.ncbi.nlm.nih.gov/pubmed/6673503?tool=bestpractice.com
Neutrophils of patients with CGD do not reduce nitroblue tetrazolium.
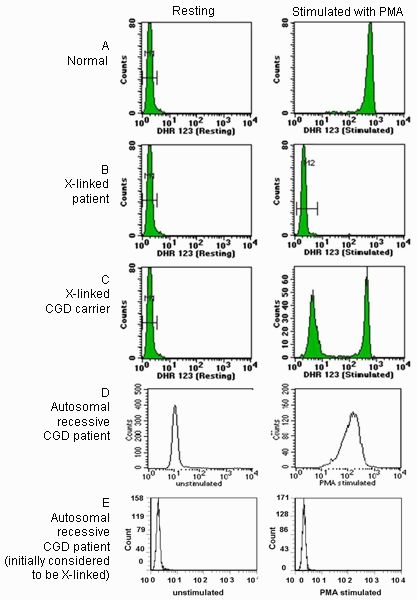
The second method, the dihydrorhodamine (DHR) 123 test, utilizes flow cytometry to detect the oxidation of dihydrorhodamine 123 in activated neutrophils. This test differentiates between X-linked and autosomal recessive forms as well.[47]Jirapongsananuruk O, Malech HL, Kuhns DB, et al. Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. J Allergy Clin Immunol. 2003 Feb;111(2):374-9.
http://www.ncbi.nlm.nih.gov/pubmed/12589359?tool=bestpractice.com
[48]Vowells SJ, Fleischer TA, Sekhsaria S, et al. Genotype-dependent variability in flow cytometric evaluation of reduced nicotinamide adenine dinucleotide phosphate oxidase function in patients with chronic granulomatous disease. J Pediatr. 1996 Jan;128(1):104-7.
http://www.ncbi.nlm.nih.gov/pubmed/8551399?tool=bestpractice.com
[49]Roesler J, Hecht M, Freihorst J, et al. Diagnosis of chronic granulomatous disease and its mode of inheritance by dihydrorhodamine 123 and flow microcytofluorometry. Eur J Pediatr. 1991 Jan;150(3):161-5.
http://www.ncbi.nlm.nih.gov/pubmed/2044584?tool=bestpractice.com
The X-linked form is typically associated with an absence of detectable oxidative burst activity, while autosomal recessive forms are usually associated with a decreased DHR shift and a wide-based histogram. Subsequent tests include Western blot or flow cytometric analysis of individual NADPH oxidase components. Genetic analysis is used to confirm the diagnosis.[50]Bonilla FA, Khan DA, Ballas ZK, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015 Nov;136(5):1186-205.e1-78.
https://www.doi.org/10.1016/j.jaci.2015.04.049
http://www.ncbi.nlm.nih.gov/pubmed/26371839?tool=bestpractice.com
If there are existing genetic test results, do not perform repeat testing unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[51]American College of Medical Genetics and Genomics. Five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2021 [internet publication].
https://web.archive.org/web/20230326143738/https://www.choosingwisely.org/societies/american-college-of-medical-genetics-and-genomics
Following the diagnosis of male patients with X-linked CGD, maternal testing is recommended.[Figure caption and citation for the preceding image starts]: Sample DHR histograms. Neutrophils were incubated with dihydrorhodamine 123 (DHR 123) and then activated with phorbol 12-myristate 13-acetate (PMA). On activation, DHR 123 is oxidized to highly fluorescent rhodamine 123 in normal neutrophils. Pre-activation histograms are shown on the left and post-activation histograms on the right. Block A shows normal response, with a large rightward shift in mean fluorescent intensity. Block B shows a patient with X-linked CGD lacking a detectable oxidative burst. Block C shows the mother of an affected patient with 2 populations of neutrophils, one normal and one with mutated gp91phox. Block D shows the typical pattern observed in patients with autosomal recessive CGD. Patients with autosomal recessive CGD can also rarely display an absence of oxidative burst activity, as shown in Block E.Permission by CCHMC clinical diagnostic immunology lab [Citation ends].