The goal is to confirm autosomal-dominant PKD (ADPKD) diagnosis and to then classify the patient’s risk of progression to kidney failure on the basis of the combination of clinical, genetic, and imaging factors.[35]Chebib FT, Torres VE. Recent advances in the management of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2018 Nov 7;13(11):1765-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6237066
http://www.ncbi.nlm.nih.gov/pubmed/30049849?tool=bestpractice.com
Extrarenal manifestations and evidence of long-term complications of ADPKD also need to be ascertained.
A positive family history of ADPKD, with signs and symptoms of renal and/or extrarenal manifestations, is highly suggestive of ADPKD. Definitive diagnosis is from imaging studies of the kidneys, or genetic testing if imaging is inconclusive.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
A likely diagnosis may be considered in patients without a positive family history, and the presence of renal cysts, with or without hepatic cysts, in the absence of other manifestations is suggestive of a different renal cystic disease.[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
However, genetic testing is indicated for a definitive diagnosis in these patients.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
At least 10% to 15% of patients with a negative family history will have a new mutation.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Appropriate counseling should be done before testing (imaging or genetic) for PKD.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
Potential discrimination, in terms of insurability and employment, associated with a positive diagnosis should be discussed.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
History
Family history may include ADPKD, end-stage renal disease (ESRD), intracranial aneurysm, hemorrhagic stroke, or subarachnoid hemorrhage. For adults with a family history of ADPKD, diagnosis may be made presymptomatically by abdominal imaging.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Common presenting symptoms include flank/abdominal pain, renal colic, and gross hematuria, and, less commonly, headaches.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
[36]Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med. 2008 Oct 2;359(14):1477-85.
http://www.ncbi.nlm.nih.gov/pubmed/18832246?tool=bestpractice.com
Urinary tract infections (UTIs) occur in 30% to 50% of adult patients with PKD.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Patients with lower tract UTIs present with a history of dysuria, urgency, and suprapubic pain. UTIs involving the renal parenchyma or cysts typically present with flank pain and fever. Patients with kidney stones may present with flank pain, hematuria, dysuria, and fever. Heartburn, reflux, nausea, dyspnea, early satiety, or increased abdominal girth may occur with severe hepatic disease.[37]Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 Mar 27;12(3):72-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097502
http://www.ncbi.nlm.nih.gov/pubmed/32231761?tool=bestpractice.com
There is an association between ADPKD and diverticulosis in the presence of ESRD.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Physical exam
Abdominal examination often reveals a palpable renal or hepatic mass. ADPKD is characterized by progressive enlargement of innumerable renal cysts and kidney size, which increases exponentially with age.[26]Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006 May 18;354(20):2122-30.
https://www.nejm.org/doi/10.1056/NEJMoa054341
http://www.ncbi.nlm.nih.gov/pubmed/16707749?tool=bestpractice.com
Kidneys become massively enlarged and may be associated with significant morbidity from the additional mass and weight.[39]Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006 Jan;1(1):148-57.
https://cjasn.asnjournals.org/content/1/1/148.long
http://www.ncbi.nlm.nih.gov/pubmed/17699202?tool=bestpractice.com
Polycystic liver disease is present in over 90% of individuals with ADPKD ages older than 35 years.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
[37]Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 Mar 27;12(3):72-83.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7097502
http://www.ncbi.nlm.nih.gov/pubmed/32231761?tool=bestpractice.com
Hepatomegaly is commonly present even before detection of liver cysts by imaging.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Hypertension is common and often occurs at a relatively young age with an average age of onset of between 30 to 34 years.[7]Gimpel C, Bergmann C, Bockenhauer D, et al. International consensus statement on the diagnosis and management of autosomal dominant polycystic kidney disease in children and young people. Nat Rev Nephrol. 2019 Nov;15(11):713-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7136168
http://www.ncbi.nlm.nih.gov/pubmed/31118499?tool=bestpractice.com
Detection of hypertension before any of the other clinical manifestations is often how the disease is first detected in young patients.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Inguinal, incisional, or paraumbilical hernias are often present.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
A cardiac murmur may be present, suggestive of mitral valve prolapse, mitral regurgitation, aortic regurgitation, or dilated aortic root.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Laboratory tests
Serum electrolytes, blood urea nitrogen, creatinine, and fasting lipid profile should be ordered initially. Creatinine can be used to estimate glomerular filtration rate.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
Elevated lipids may be associated with a higher likelihood of progressive renal insufficiency.[40]Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011 Mar;6(3):640-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3082424
http://www.ncbi.nlm.nih.gov/pubmed/21088290?tool=bestpractice.com
Urinalysis should be ordered in all patients to detect presence of increased urinary albumin excretion or proteinuria. If increased urinary albumin excretion or proteinuria are found, this indicates a higher likelihood of progression to chronic kidney disease.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
[40]Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011 Mar;6(3):640-7.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3082424
http://www.ncbi.nlm.nih.gov/pubmed/21088290?tool=bestpractice.com
Microscopic or macroscopic hematuria is common. Leukocyturia can be seen in people with PKD, but it does not always indicate UTI, and a urine culture should be obtained in that situation. Urine culture should always be obtained at initial evaluation if there are symptoms of UTI or fever.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Urine culture may be negative even with serious urine infection, because cysts do not communicate with the urinary tract. In patients with abdominal or flank pain and/or tenderness and fever, a C-reactive protein should be requested.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
All patients with metabolically active stone disease should have 24-hour urine collection for urine biochemistry (urine volume, pH, oxalate, uric acid, citrate, phosphate, sodium, and calcium, as well as creatinine to assess the completeness of the collection), and supersaturation should be calculated. Low urine citrate and urine pH are the main metabolic factors predisposing to stone formation in ADPKD.[41]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.
http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com
Renal imaging
Imaging shows the presence of renal cysts with, or without, hepatic cysts. Ultrasonography is the most common initial test to order because of low cost, wide availability, and safety.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
Unified ultrasound diagnostic criteria for the diagnosis of ADPKD in at-risk people (those from families with ADPKD of unknown genotype) have been developed.[42]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2615723
http://www.ncbi.nlm.nih.gov/pubmed/18945943?tool=bestpractice.com
The presence of at least three (unilateral or bilateral) renal cysts and two cysts in each kidney is sufficient for diagnosis of at-risk individuals ages 15 to 39 and 40 to 59 years, respectively; in those over 60 years, four or more cysts in each kidney is required.[42]Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2615723
http://www.ncbi.nlm.nih.gov/pubmed/18945943?tool=bestpractice.com
These criteria should not be applied to magnetic resonance imaging (MRI) or computed tomography (CT) scans as could lead to false-positive results.
Contrast-enhanced CT scan or MRI detect cysts of 2 to 3 mm in diameter and are particularly useful for diagnosis in younger patients.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
In patients at risk of ADPKD younger than 30 years of age, a test criterion of a total of >10 renal cysts seen on MRI is considered sufficient for diagnosis.[43]Pei Y, Hwang YH, Conklin J, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015 Mar;26(3):746-53.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4341484
http://www.ncbi.nlm.nih.gov/pubmed/25074509?tool=bestpractice.com
The use of CT needs to be weighed against the radiation dose.
Imaging by CT scan or MRI should be part of the initial evaluation of ADPKD, as it provides precise, standardized measurement of maximum kidney length, width, and depth measurements, and an estimate of total kidney volume.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
These quantitative data are of prognostic value.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
An image-based classification system based on total kidney volumes from CT scan or MRI has been used to identify potential cases of more rapidly progressive disease. This classification system optimizes patient selection for specific disease therapy.[44]Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015 Jan;26(1):160-72.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4279733
http://www.ncbi.nlm.nih.gov/pubmed/24904092?tool=bestpractice.com
In patients with a negative family history, imaging-based diagnosis is not sufficient because criteria were developed in individuals who had a 50% risk of ADPKD.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
A likely diagnosis of ADPKD can be considered if, despite no family history, there are >10 cysts in each kidney, and there is no evidence of renal or extrarenal manifestations of another cystic renal disease that could explain the phenotype; however, genetic testing is indicated to confirm diagnosis.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[11]Suwabe T, Shukoor S, Chamberlain AM, et al. Epidemiology of autosomal dominant polycystic kidney disease in Olmsted County. Clin J Am Soc Nephrol. 2020 Jan 7;15(1):69-79.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6946081
http://www.ncbi.nlm.nih.gov/pubmed/31791998?tool=bestpractice.com
A positron emission tomography scan is more sensitive than CT or MRI for detecting cyst infections.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: CT scan of abdomen and pelvis of patient with mild diseaseFrom collection of Dr M. Hogan [Citation ends].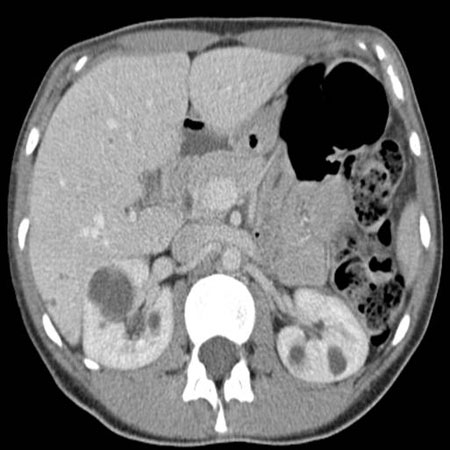 [Figure caption and citation for the preceding image starts]: MRI of abdomen and pelvis of patient with symptomatic polycystic kidney diseaseFrom collection of Dr M. Hogan [Citation ends].
[Figure caption and citation for the preceding image starts]: MRI of abdomen and pelvis of patient with symptomatic polycystic kidney diseaseFrom collection of Dr M. Hogan [Citation ends].
Investigation of extrarenal manifestations
Cardiovascular abnormalities include hypertension, left ventricular hypertrophy, aortic root dilatation, arterial aneurysms, heart valve abnormalities, and intracranial aneurysms.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
ECG and echocardiogram are used for the initial assessment of cardiovascular complications such as left ventricular hypertrophy (can be present without hypertension), cardiac function (e.g., ejection fraction, diastolic dysfunction), and valvular abnormalities.[45]Kuo IY, Chapman AB. Polycystins, ADPKD, and cardiovascular disease. Kidney Int Rep. 2020 Apr;5(4):396-406.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7136326
http://www.ncbi.nlm.nih.gov/pubmed/32274448?tool=bestpractice.com
CT scan and MRI may also provide evidence of extrarenal cysts. Hepatic cysts are the most common extrarenal manifestation.[1]Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6592047
http://www.ncbi.nlm.nih.gov/pubmed/30523303?tool=bestpractice.com
Pancreatic cysts (prevalence 9% on ultrasound screening, 36% on MRI) are almost always asymptomatic with rare occurrences of recurrent pancreatitis.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Pancreatic cysts are rarely associated with intraductal papillary mucinous tumor or carcinoma.[38]Mikolajczyk AE, Te HS, Chapman AB. Gastrointestinal manifestations of autosomal-dominant polycystic kidney disease. Clin Gastroenterol Hepatol. 2017 Jan;15(1):17-24.
https://www.cghjournal.org/article/S1542-3565(16)30364-0/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/27374006?tool=bestpractice.com
Additionally, if nephrolithiasis is suspected, CT or MRI imaging can differentiate stone disease from cyst wall calcification or parenchymal calcifications. About 20% of patients have kidney stones, and the composition of these is typically uric acid or calcium oxalate.[41]Torres VE, Erickson SB, Smith LH, et al. The association of nephrolithiasis and autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1988 Apr;11(4):318-25.
http://www.ncbi.nlm.nih.gov/pubmed/3354568?tool=bestpractice.com
[46]Grampsas SA, Chandhoke PS, Fan J, et al. Anatomic and metabolic risk factors for nephrolithiasis in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2000 Jul;36(1):53-7.
http://www.ncbi.nlm.nih.gov/pubmed/10873872?tool=bestpractice.com
Dual-energy CT is a useful method to discriminate uric acid stones from other (non-uric acid) renal stones.[31]Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015 Jul;88(1):17-27.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4913350
http://www.ncbi.nlm.nih.gov/pubmed/25786098?tool=bestpractice.com
The presence of intracranial aneurysms is four times higher in patients with ADPKD than in the general population.[4]Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35.
http://www.ncbi.nlm.nih.gov/pubmed/30819518?tool=bestpractice.com
Identification of sentinel headaches and prompt diagnosis and treatment of a leaking aneurysm is an important determinant of outcome.[34]Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2002 Jan;13(1):269-76.
https://jasn.asnjournals.org/content/13/1/269.long
http://www.ncbi.nlm.nih.gov/pubmed/11752048?tool=bestpractice.com
If subarachnoid hemorrhage is suspected, a noncontrast head CT is performed, which, if nondiagnostic, should be followed by lumbar puncture.[47]Hoh BL, Ko NU, Amin-Hanjani S, et al. 2023 Guideline for the management of patients with aneurysmal subarachnoid hemorrhage: a guideline from the American Heart Association/American Stroke Association. Stroke. 2023 Jul;54(7):e314-70.
https://www.ahajournals.org/doi/full/10.1161/STR.0000000000000436?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/37212182?tool=bestpractice.com
If these tests are positive, the patient should be admitted immediately to a neurosurgical intensive care unit for vessel imaging, usually a CT angiogram, and treatment of the leaking intracranial aneurysm.[48]Chung DY, Abdalkader M, Nguyen TN. Aneurysmal subarachnoid hemorrhage. Neurol Clin. 2021 May;39(2):419-42.
http://www.ncbi.nlm.nih.gov/pubmed/33896527?tool=bestpractice.com
See our topics Cerebral aneurysm and Subarachnoid hemorrhage.
Genetic testing
Genetic testing can be used in the following cases:[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
The imaging results are equivocal or inconclusive
To confirm a presumed diagnosis in the absence of family history of the disease (conclusive proof of the diagnosis in these patients relies on mutation analysis)
When a definite diagnosis is required in a younger patient, such as a potential living related kidney donor
For prenatal and preimplantation genetic testing.
Mutations in the PKD1 gene cause more severe disease than those in the PKD2 gene.[2]Lavu S, Vaughan LE, Senum SR, et al. The value of genotypic and imaging information to predict functional and structural outcomes in ADPKD. JCI Insight. 2020 Aug 6;5(15):e138724.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7455088
http://www.ncbi.nlm.nih.gov/pubmed/32634120?tool=bestpractice.com
Genetic testing for PKD1 mutation screening is expensive and challenging due to its large size and complexity.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
It can be done by linkage or sequence analysis; however, sequence analysis is preferred. Despite comprehensive screening, 10% to 15% of patients suspicious of ADPKD have no mutation detected in either PKD1 or PKD2.[21]Lanktree MB, Haghighi A, di Bari I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies. Clin J Am Soc Nephrol. 2021 May 8;16(5):790-9.
https://cjasn.asnjournals.org/content/16/5/790.long
http://www.ncbi.nlm.nih.gov/pubmed/32690722?tool=bestpractice.com
Patients can be rescreened for mutations of a newly identified gene for ADPKD, GANAB, and somatic mosaicism.[20]Iliuta IA, Kalatharan V, Wang K, et al. Polycystic kidney disease without an apparent family history. J Am Soc Nephrol. 2017 Sep;28(9):2768-76.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5576926
http://www.ncbi.nlm.nih.gov/pubmed/28522688?tool=bestpractice.com
If there are existing genetic test results, do not order a duplicate test unless there is uncertainty about the existing result, e.g., the result is inconsistent with the patient’s clinical presentation or the test methodology has changed.[55]American College of Medical Genetics and Genomics. Five things physicians and patients should question. Choosing Wisely, an initiative of the ABIM Foundation. 2021 [internet publication].
https://web.archive.org/web/20230326143738/https://www.choosingwisely.org/societies/american-college-of-medical-genetics-and-genomics
All published mutations and other variants are publicly available in the ADPKD variant database.
PKD Foundation: ADPKD variant database
Opens in new window If a mutation is known in one family member, this mutation can be confirmed in other family members at a considerably lower cost.
An emerging approach is whole-exome sequencing. In one validation study, whole-genome sequencing was able to overcome pseudogene homology and identify all types of variants in the PKD1 gene.[19]Mallawaarachchi AC, Lundie B, Hort Y, et al. Genomic diagnostics in polycystic kidney disease: an assessment of real-world use of whole-genome sequencing. Eur J Hum Genet. 2021 May;29(5):760-70.
http://www.ncbi.nlm.nih.gov/pubmed/33437033?tool=bestpractice.com
